×
Close
註冊
登入
主页
用户
博客
照片
视频
医学图书S馆
医学图书M馆
医学图书D馆
医学术语
群组
活动
用户工具
用户指南
问答
谁看过我
反馈
语言
English
中文
推广
×
Close
CN1699 Our Business - Guide for Agents
分类
Biochemistry & Molecular Biology
37484
Global Medical University
6716
Allergy
1956
Anatomy & Morphology
1796
Andrology
414
Anesthesia & Intensive Care
1533
Anesthesiology
6648
Audiology & Speech-Language Pathology
393
Behavioral Sciences
111
Biochemical Research Methods
9135
Biodiversity Conservation
423
Biology
11203
Biophysics
12801
Biotechnology & Applied Microbiology
10760
Cardiac & Cardiovascular Systems
38211
Cardiovascular & Respiratory Systems
1855
Cell & Tissue Engineering
866
Cell Biology
13306
Chemistry, Analytical
4979
Chemistry, Applied
14031
Chemistry, Medicinal
10506
Chemistry, Multidisciplinary
22895
Clinical Immunology & Infectious Disease
543
Clinical Medicine
10601
Clinical Neurology
20019
Clinical Psychology & Psychiatry
1946
Critical Care Medicine
3998
Dentistry, Oral Surgery & Medicine
17118
Dermatology
8190
Developmental Biology
8041
Ecology
676
Education, Scientific Disciplines
2481
Emergency Medicine
5291
Endocrinology, Metabolism & Nutrition
30687
Engineering, Biomedical
4679
Entomology
537
Environmental Medicine & Public Health
6159
Evolutionary Biology
321
Gastroenterology & Hepatology
14940
General & Internal Medicine
8808
Genetics & Heredity
19133
Geriatrics & Gerontology
6105
Gerontology
390
Health Care Sciences & Services
20149
Health Policy & Services
747
Hematology
6450
Immunology
31938
Infectious Diseases
17246
Integrative & Complementary Medicine
3644
Medical Ethics
1420
Medical Informatics
3252
Medical Laboratory Technology
433
Medicine, General & Internal
52118
Medicine, Legal
671
Medicine, Research & Experimental
23666
Microbiology
29719
Mycology
0
Nanoscience & Nanotechnology
6893
Neuroimaging
1732
Neurology
6153
Neurosciences
49840
Nursing
11401
Nutrition & Dietetics
9232
Obstetrics & Gynecology
10046
Oncology
65559
Ophthalmology
12603
Optics
6161
Orthopedics
14032
Orthopedics, Rehabilitation & Sports Medicine
2292
Otolaryngology
1871
Otorhinolaryngology
5935
Parasitology
1274
Pathology
6104
Pediatrics
26226
Peripheral Vascular Disease
5806
Pharmacology & Pharmacy
46504
Pharmacology/Toxicology
15032
Physiology
10476
Polymer Science
820
Primary Health Care
1001
Psychiatry
25712
Psychology
6162
Psychology, Applied
131
Psychology, Biological
482
Psychology, Clinical
1001
Psychology, Developmental
241
Psychology, Educational
176
Psychology, Experimental
187
Psychology, Mathematical
0
Psychology, Multidisciplinary
1780
Psychology, Psychoanalysis
41
Psychology, Social
132
Public Health & Health Care Science
2525
Public, Environmental & Occupational Health
33267
Quantum Science & Technology
0
Radiology, Nuclear Medicine & Imaging
15634
Radiology, Nuclear Medicine & Medical Imaging
9929
Rehabilitation
3445
Remote Sensing
0
Reproductive Biology
3786
Reproductive Medicine
1502
Research/Laboratory Medicine & Medical Technology
5129
Respiratory System
9146
Rheumatology
7443
Social Sciences, Biomedical
1431
Substance Abuse
3241
Surgery
42248
Toxicology
5330
Transplantation
977
Tropical Medicine
314
Urology & Nephrology
16396
Veterinary Sciences
35
Virology
2902
Zoology
0
渠道
JOURNAL OF COMPUTER-AIDED MOLECULAR DESIGN
196
AMERICAN JOURNAL OF PHYSIOLOGY-ENDOCRINOLOGY AND METABOLISM
223
ANTIOXIDANTS
2594
BIOMOLECULES
2187
BIOORGANIC CHEMISTRY
2385
CELL AND BIOSCIENCE
642
CELLULAR & MOLECULAR BIOLOGY LETTERS
384
CELLULAR AND MOLECULAR LIFE SCIENCES
1657
COLLOIDS AND SURFACES B-BIOINTERFACES
2090
COMPUTATIONAL AND STRUCTURAL BIOTECHNOLOGY JOURNAL
1668
CYTOKINE
941
CYTOKINE & GROWTH FACTOR REVIEWS
200
FREE RADICAL BIOLOGY AND MEDICINE
1506
GENOME RESEARCH
622
INTERNATIONAL JOURNAL OF BIOLOGICAL SCIENCES
20
INTERNATIONAL REVIEW OF CELL AND MOLECULAR BIOLOGY
346
JOURNAL OF BIOLOGICAL CHEMISTRY
3309
JOURNAL OF CELLULAR BIOCHEMISTRY
110
JOURNAL OF GENETICS AND GENOMICS
647
JOURNAL OF INORGANIC BIOCHEMISTRY
912
JOURNAL OF INTEGRATIVE PLANT BIOLOGY
101
JOURNAL OF LIPID RESEARCH
33
JOURNAL OF MOLECULAR BIOLOGY
1016
JOURNAL OF PHOTOCHEMISTRY AND PHOTOBIOLOGY B-BIOLOGY
552
MOLECULAR BIOLOGY
328
MOLECULAR CARCINOGENESIS
63
NATURE CHEMICAL BIOLOGY
923
NATURE PROTOCOLS
555
NEUROCHEMISTRY INTERNATIONAL
539
PROGRESS IN LIPID RESEARCH
0
REDOX BIOLOGY
1
SCIENCE SIGNALING
181
SIGNAL TRANSDUCTION AND TARGETED THERAPY
1361
BIOCHEMISTRY AND CELL BIOLOGY
146
BIOMEDICAL JOURNAL
0
BIOORGANIC & MEDICINAL CHEMISTRY
1
BIOPHYSICAL CHEMISTRY
0
BIOPOLYMERS
28
BIOTECHNIQUES
406
BMC BIOCHEMISTRY
155
CELL BIOCHEMISTRY AND FUNCTION
55
CHEMBIOCHEM
352
CHEMICAL BIOLOGY & DRUG DESIGN
164
CURRENT ISSUES IN MOLECULAR BIOLOGY
565
FRONTIERS IN MOLECULAR BIOSCIENCES
3202
JOURNAL OF FOOD BIOCHEMISTRY
259
JOURNAL OF MOLECULAR RECOGNITION
35
LIPIDS
23
NEUROCHEMICAL RESEARCH
860
PHYSICAL BIOLOGY
129
PROGRESS IN BIOPHYSICS & MOLECULAR BIOLOGY
6
PROTEIN SCIENCE
152
LIPIDS IN HEALTH AND DISEASE
768
VITAMINS AND HORMONES
298
BIOCHEMISTRY INSIGHTS
8
BIOCHEMISTRY MOSCOW-SUPPLEMENT SERIES B-BIOMEDICAL CHEMISTRY
97
INDIAN JOURNAL OF CLINICAL BIOCHEMISTRY
274
PROTEOMES
63
MOLECULAR BIOLOGY RESEARCH COMMUNICATIONS
91
REVIEWS OF PHYSIOLOGY BIOCHEMISTRY AND PHARMACOLOGY
64
GENES & GENOMICS
495
INTERNATIONAL JOURNAL OF PEPTIDE RESEARCH AND THERAPEUTICS
399
JOURNAL OF BIOMOLECULAR NMR
97
SCI时时刷
search
全部
推荐
+
In silico design of dehydrophenylalanine containing peptide activators of glucokinase using pharmacophore modelling, molecular dynamics and machine learning: implications in type 2 diabetes
Diabetes represents a significant global health challenge associated with substantial healthcare costs and therapeutic com...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
ConoDL: a deep learning framework for rapid generation and prediction of conotoxins
Conotoxins, being small disulfide-rich and bioactive peptides, manifest notable pharmacological potential and find extensi...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
MolGraph: a Python package for the implementation of molecular graphs and graph neural networks with TensorFlow and Keras
Molecular machine learning (ML) has proven important for tackling various molecular problems, such as predicting molecular...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Combining crystallographic and binding affinity data towards a novel dataset of small molecule overlays
Although small molecule superposition is a standard technique in drug discovery, a rigorous performance assessment of the ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Promoter recognition specificity of Corynebacterium glutamicum stress response sigma factors σD and σH deciphered using computer modeling and point mutagenesis
This study aimed to reveal interactions of the stress response sigma subunits (factors) σD and σH of RNA polymer...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Understanding the relationship between preferential interactions of peptides in water-acetonitrile mixtures with protein-solvent contact surface area
The influence of polar, water-miscible organic solvents (POS) on protein structure, stability, and functional activity is ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Identification of novel inhibitors targeting PI3Kα via ensemble-based virtual screening method, biological evaluation and molecular dynamics simulation
PIK3CA gene encoding PI3K p110α is one of the most frequently mutated and overexpressed in majority of human cancers....
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Steered molecular dynamics simulation as a post-process to optimize the iBRAB-designed Fab model
Therapeutic monoclonal antibodies are an effective method of treating acute infectious diseases. However, knowing which of...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Comparative assessment of physics-based in silico methods to calculate relative solubilities
Relative solubilities, i.e. whether a given molecule is more soluble in one solvent compared to others, is a critical para...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Computational Identification and Illustrative Standard for Representation of Unimolecular G-Quadruplex Secondary Structures (CIIS-GQ)
G-quadruplexes refer to a large group of nucleic acid–based structures. In recent years, they have been attracting a...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Structure-based pose prediction: Non-cognate docking extended to macrocyclic ligands
So-called “cross-docking” is the prediction of the bound configuration of small-molecule ligands that differ f...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
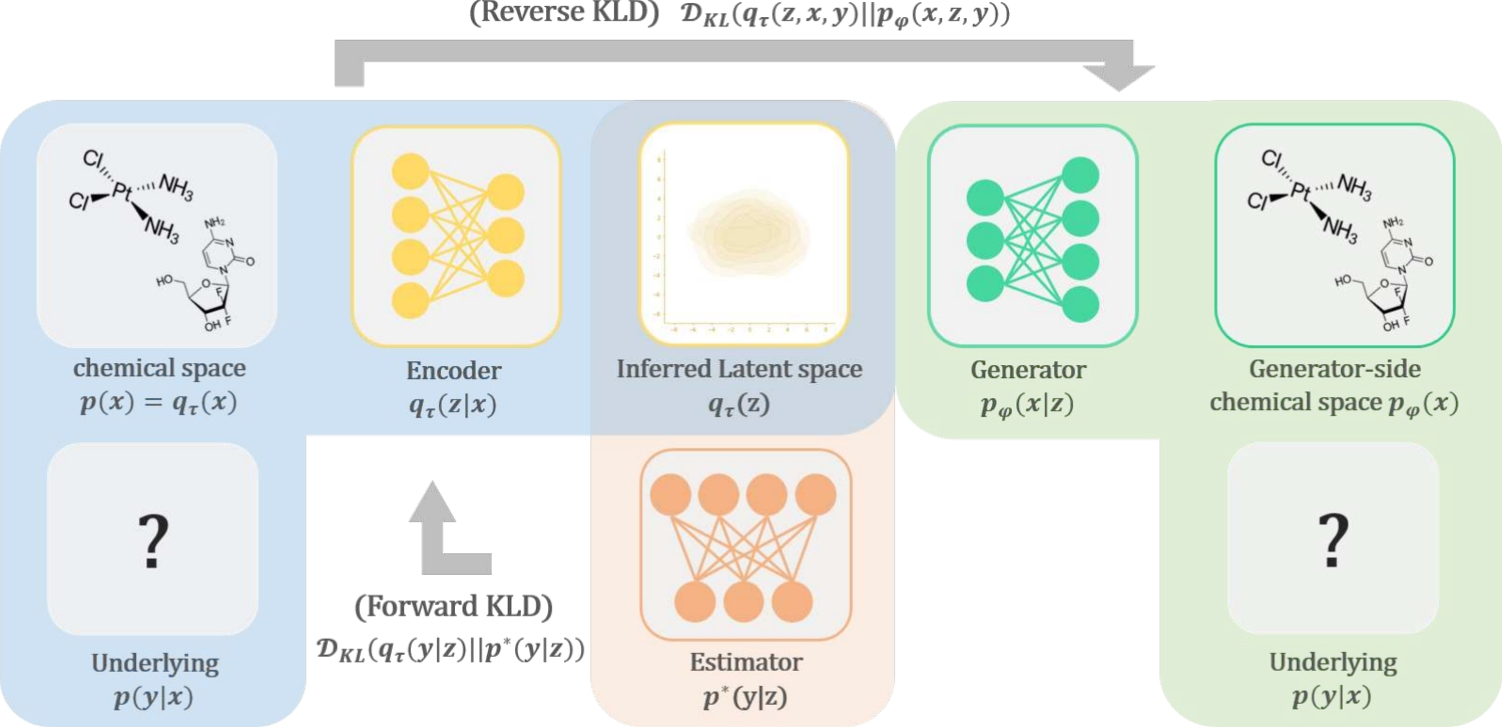
De novo drug design through gradient-based regularized search in information-theoretically controlled latent space
Over the last decade, automatic chemical design frameworks for discovering molecules with drug-like properties have signif...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Computational design and experimental confirmation of a disulfide-stapled YAP helixα1-trap derived from TEAD4 helical hairpin to selectively capture YAP α1-helix with potent antitumor activity
Human Hippo signaling pathway is an evolutionarily conserved regulator network that controls organ development and has bee...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
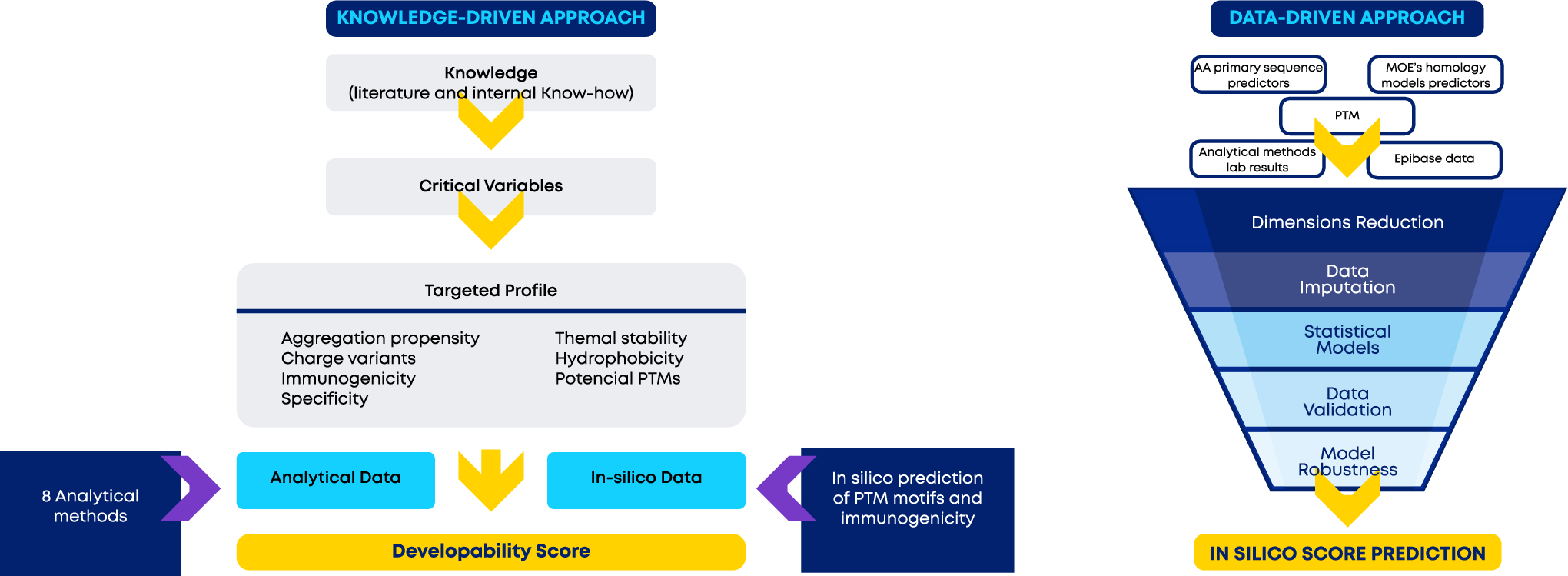
Holistic in silico developability assessment of novel classes of small proteins using publicly available sequence-based predictors
The development of novel therapeutic proteins is a lengthy and costly process, with an average attrition rate of 91% (Thom...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
FitScore: a fast machine learning-based score for 3D virtual screening enrichment
Enhancing virtual screening enrichment has become an urgent problem in computational chemistry, driven by increasingly lar...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
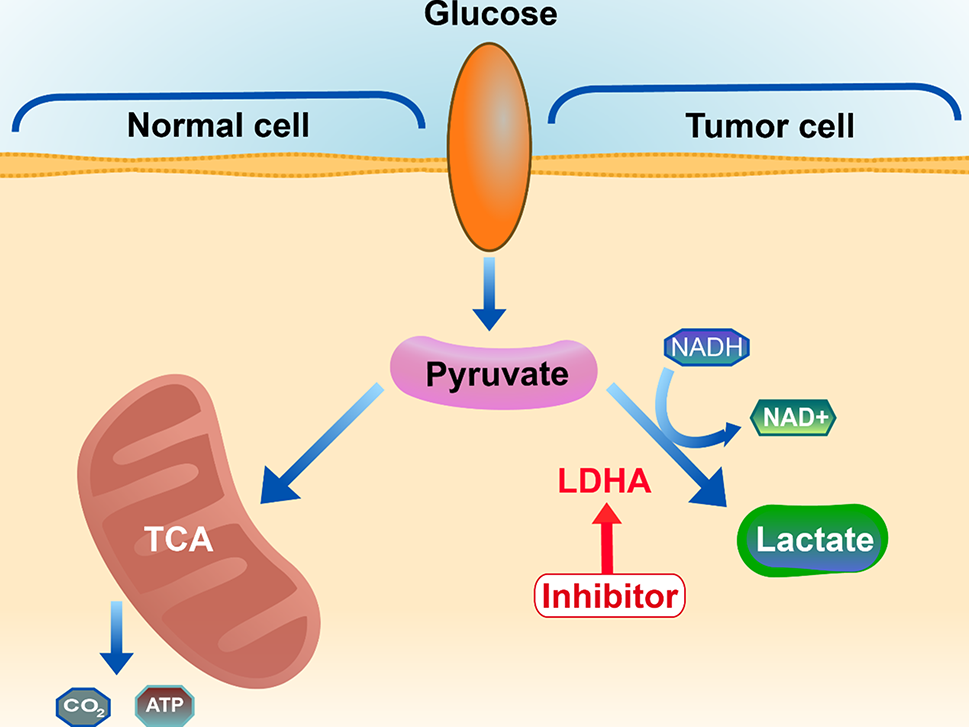
Development of human lactate dehydrogenase a inhibitors: high-throughput screening, molecular dynamics simulation and enzyme activity assay
Lactate dehydrogenase A (LDHA) is highly expressed in many tumor cells and promotes the conversion of pyruvate to lactic a...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
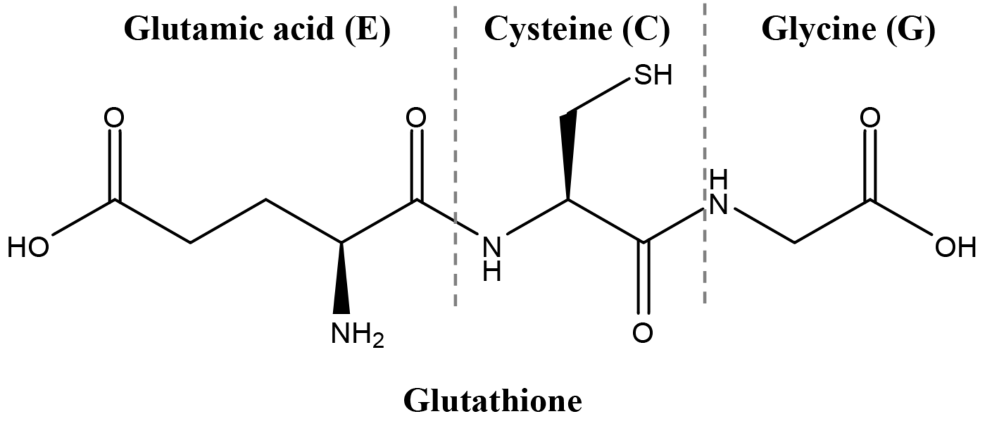
Development of QSARs for cysteine-containing di- and tripeptides with antioxidant activity:influence of the cysteine position
Antioxidants agents play an essential role in the food industry for improving the oxidative stability of food products. In...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
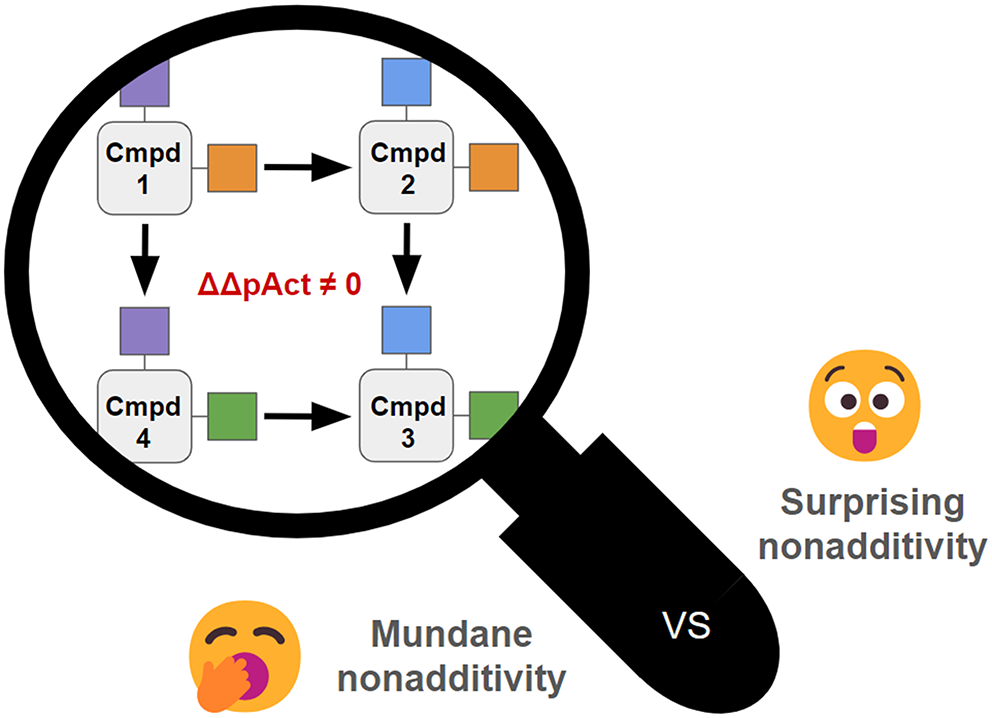
From mundane to surprising nonadditivity: drivers and impact on ML models
Nonadditivity (NA) in Structure-Activity and Structure-Property Relationship (SAR) data is a rare but very information ric...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
MDFit: automated molecular simulations workflow enables high throughput assessment of ligands-protein dynamics
Molecular dynamics (MD) simulation is a powerful tool for characterizing ligand–protein conformational dynamics and ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Structural impacts of two disease-linked ADAR1 mutants: a molecular dynamics study
Adenosine deaminases acting on RNA (ADARs) are pivotal RNA-editing enzymes responsible for converting adenosine to inosine...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
User-centric design of a 3D search interface for protein-ligand complexes
In this work, we present the frontend of GeoMine and showcase its application, focusing on the new features of its latest ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
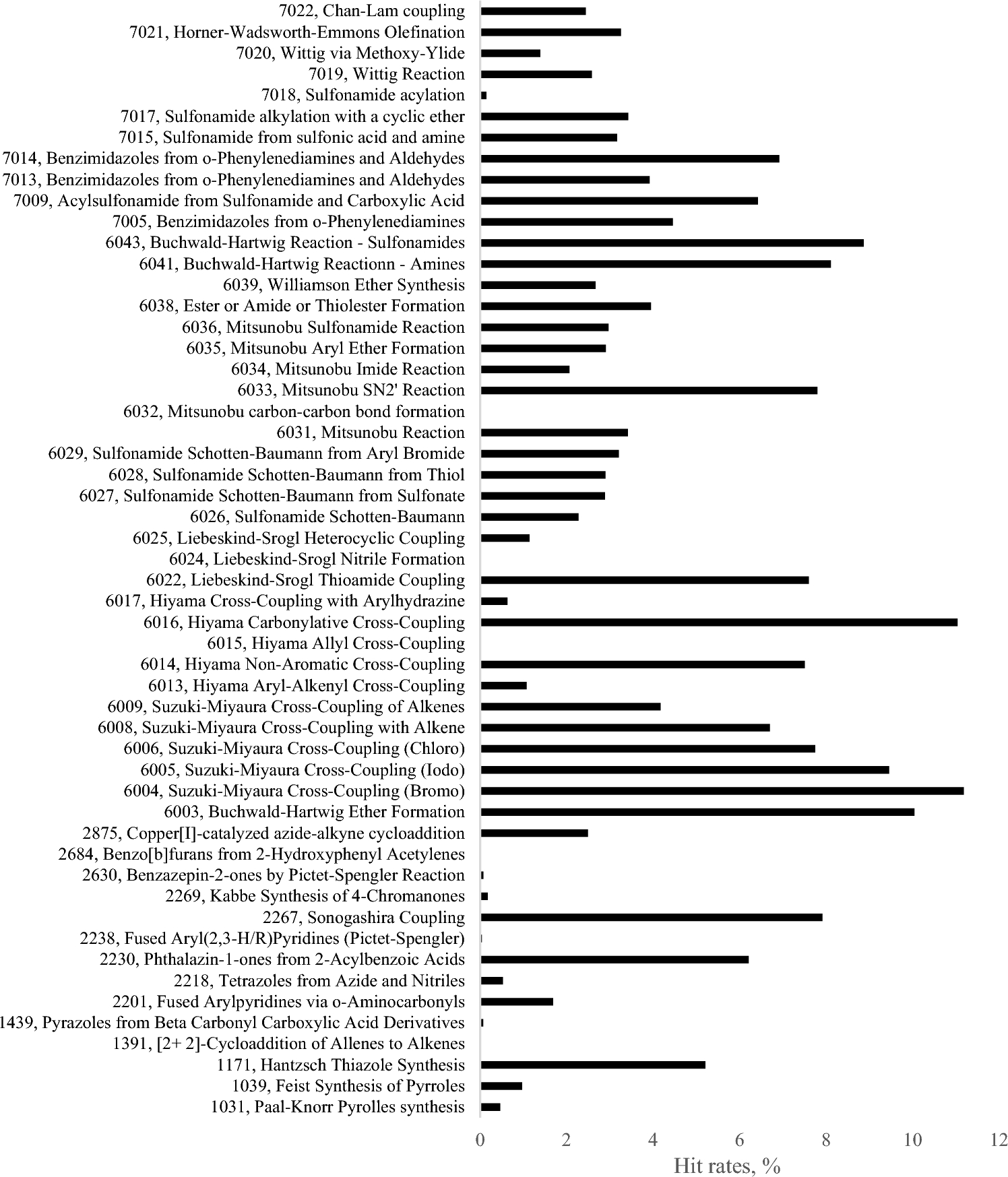
Correlation of protein binding pocket properties with hits’ chemistries used in generation of ultra-large virtual libraries
Although the size of virtual libraries of synthesizable compounds is growing rapidly, we are still enumerating only tiny f...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Reactivities of acrylamide warheads toward cysteine targets: a QM/ML approach to covalent inhibitor design
Covalent inhibition offers many advantages over non-covalent inhibition, but covalent warhead reactivity must be carefully...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
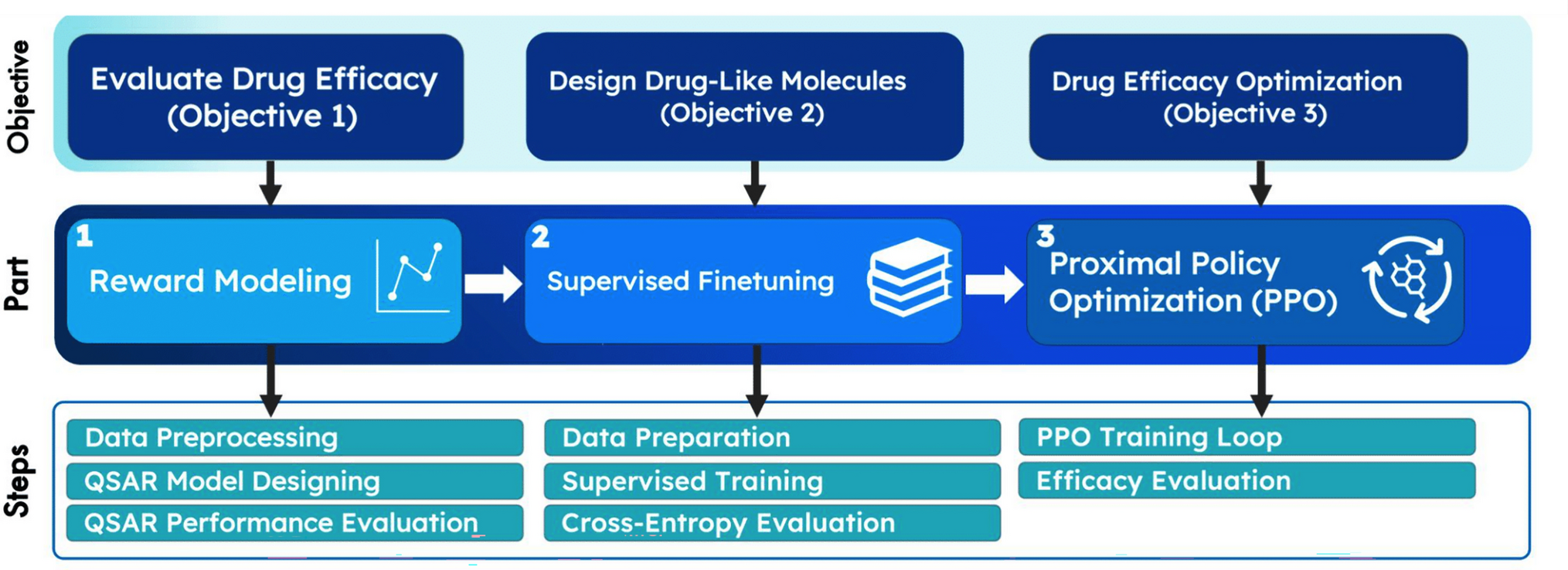
De novo drug design as GPT language modeling: large chemistry models with supervised and reinforcement learning
In recent years, generative machine learning algorithms have been successful in designing innovative drug-like molecules. ...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
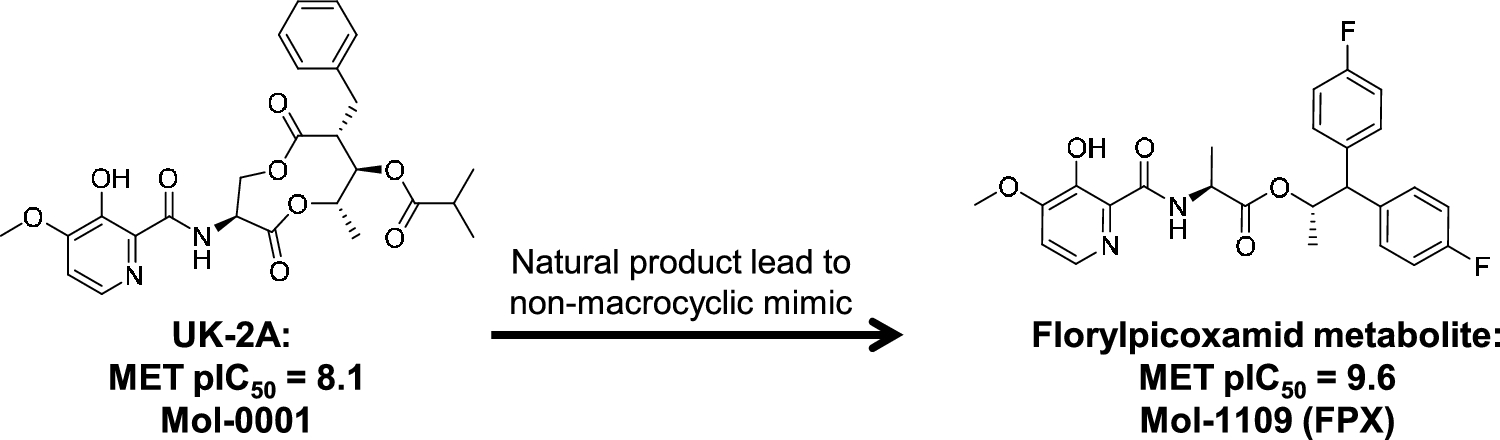
From UK-2A to florylpicoxamid: Active learning to identify a mimic of a macrocyclic natural product
Scaffold replacement as part of an optimization process that requires maintenance of potency, desirable biodistribution, m...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
On the relevance of query definition in the performance of 3D ligand-based virtual screening
Ligand-based virtual screening (LBVS) methods are widely used to explore the vast chemical space in the search of novel co...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Computational peptide discovery with a genetic programming approach
The development of peptides for therapeutic targets or biomarkers for disease diagnosis is a challenging task in protein e...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Identifying and characterising promising small molecule inhibitors of kinesin spindle protein using ligand-based virtual screening, molecular docking, molecular dynamics and MM‑GBSA calculations
The kinesin spindle protein (Eg5) is a mitotic protein that plays an essential role in the formation of the bipolar spindl...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
Benchmarking ANI potentials as a rescoring function and screening FDA drugs for SARS-CoV-2 Mpro
Here, we introduce the use of ANI-ML potentials as a rescoring function in the host–guest interaction in molecular d...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
The AI-driven Drug Design (AIDD) platform: an interactive multi-parameter optimization system integrating molecular evolution with physiologically based pharmacokinetic simulations
Computer-aided drug design has advanced rapidly in recent years, and multiple instances of in silico designed molecules ad...
Journal Of Computer-aided Molecular Design
comment
0
thumb_up
0
阅读更多
Modal title
×
Modal title
×
分享
登入
Global News and Health Forum
Join Now!
馬上登入
記住我
忘記密碼?
或者使用
Linkedin