
記住我
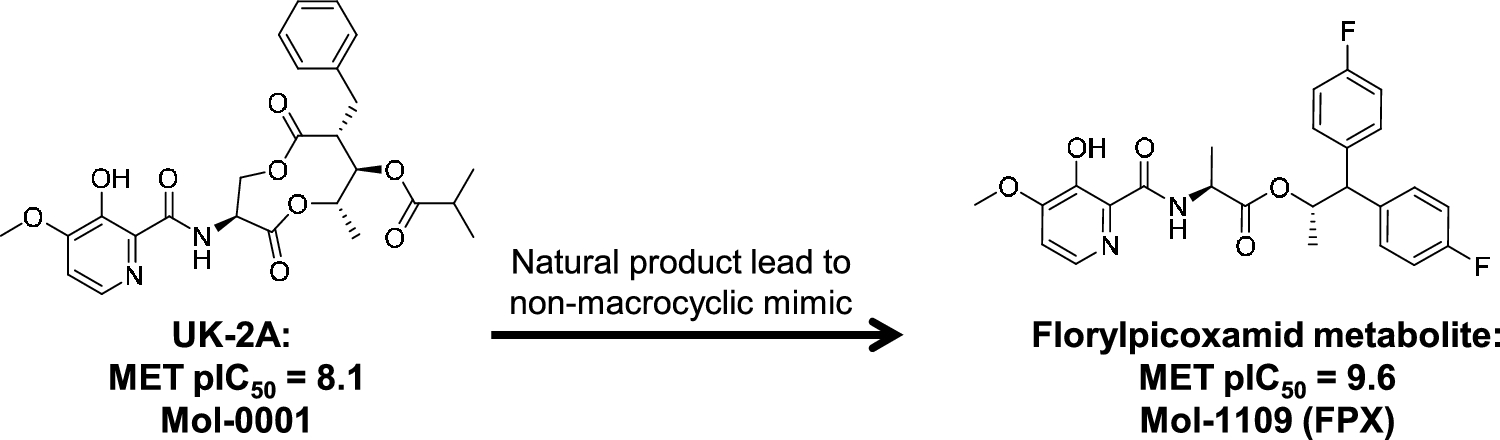





We report results for iterative model refinement leading from the natural product antifungal UK-2A to FPX, beginning with a systematic procedure for identifying an informative multiple-ligand alignment and then proceeding through multiple rounds of QuanSA model refinement using an active learning strategy. We also detail a method to generate non-macrocyclic candidate compounds using very sparse data by combining virtual-screening-based central scaffold replacement with a simple method to “staple” appropriate substituents onto the replacement scaffolds.
Fig. 3
Procedure for identifying a high-quality ligand-based binding site hypothesis
Initial multiple-ligand alignmentThe QuanSA methodology derives a pocket-field beginning from an initial mutual alignment of a set of training ligands [11, 12], where each ligand has multiple possible initial poses. When protein structure information is available, it is possible to make use of the experimentally determined relative poses of prior known bound ligands in order to guide the construction of the initial set of training poses. Here, no such suitable protein co-crystal structure existed. Rather than using crystallographic data, it is also possible to make use of a carefully constructed multiple ligand alignment to guide model-building. In cases where scaffold diversity exists among highly active molecules, such alignments can provide significant constraints on the overall ligand alignment problem.
Here, the initial set of active project compounds contained significant diversity, both within the central macrocycle as well as in the pendant functionality. Figure 3 shows the procedure used to identify a high-quality ligand-based binding site hypothesis using only the data from the earliest set of synthesized molecules. There are two key ideas: (1) to identify structurally diverse active ligands from which to produce multiple ligands alignments; and (2) to select which of the alternative hypotheses of relative bound poses is quantitatively the best. The 30 molecules from within the top 1.0 log unit of experimentally determined activity among the training molecules were used as input to identify the four most 2D structurally diverse compounds (molecules 13, 89, 2, and 64 in Fig. 3). They were selected automatically based on 2D dissimilarity (see the “Methods and data” section for details).
These molecules (to the right of UK-2A in Fig. 3) differed in terms of size and flexibility within the central macrocycle as well as the composition of the right-hand substituents. They were used, with the addition of UK-2A, as input to the the multiple-ligand alignment functionality of the eSim method [21], which resulted in several alternative mutual superimpositions. In order to assess which mutual alignment was most likely to reflect the true relative poses of the molecules, the alternative alignments were ranked based on their ability to separate highly active molecules from relatively inactive ones within the initial 100-molecule training set. The chosen hypothesis shown in Fig. 3 was able to distinguish highly active (pIC\(_ \ge 8.5\)) from less active (pIC\(_ \le 7.5\)) compounds with an ROC Area of 0.92. The 3D joint superimposition shows the tight alignment of the common left-hand moiety (the “warhead”) with the variation in the macrocycle and right-hand elements of the molecules.
Fig. 4
Example molecules chosen for model refinement in successive rounds of QuanSA testing and refinement
Iterative model refinementThe chosen multiple-ligand alignment from Fig. 3 was used to guide construction of the initial QuanSA model pocket-field. The method allows for incremental iterative refinement based on the availability of new structure-activity data. Figure 4 shows examples of molecules automatically selected by QuanSA for model refinement based on expectations of high activity (left side) or based on expectations of being informative (right side) through multiple rounds of compound selection and model refinement. Intuitively, selection of candidate molecules based on predictions of high activity is an obvious strategy. In an active-learning paradigm, one also seeks to identify maximally informative molecules [22]. One representative example of each type of selection is shown for each of the first four rounds.
The process of scoring candidate molecules in a QuanSA pocket-field results in a prediction of activity and bound pose, along with a number of prediction quality metrics. The novelty metric characterizes the degree to which a candidate molecule is well-covered by the current set of training molecules. Candidate molecule predictions also indicate which training molecule was the nearest-neighbor (NN) in a 3D molecular similarity calculation based on the predicted bound pose.
Here, in each round, the 200 least novel (i.e. best covered) predicted candidate molecules were identified, and, of this subset, the top ten with highest predicted activity were selected for model refinement (see left-hand examples from Fig. 4). The maximally informative set of ten for each round captured a group of molecules that could be thought of as having unexpectedly low activity. Informative molecules were identified from the subset whose NN training molecule similarity was high (top 100 highest NN similarity or NN similarity \(\ge \) 0.85) and where the NN training molecule’s activity was also high (pIC\(_ \ge 8.5\)). From that subset, the ten molecules with the lowest predicted activity were selected (see right-hand examples from Fig. 4).
Fig. 5
Selections of active (left, predicted poses in salmon carbons) and informative molecules (right, yellow carbons) for Round-00 shown against the predicted pose of UK-2A
In the early rounds, the compounds predicted to be highly active all had a central macrocyclic scaffold that was found among the most highly active training compounds, as would be expected given the starting point of lead optimization. However, after three rounds of model refinement (a cumulative addition of 60 molecules to the original model), a non-macrocycle was correctly identified and chosen as a highly active molecule (Fig. 4, lower left).
In contrast, the compounds predicted to be maximally informative included non-macrocycles even in the initial round of candidate selection. These compounds were deemed to be information rich: the predicted activities were low, yet these candidate molecules had very high 3D similarity to highly active train compounds. Model evolution through inclusion of these informative compounds broadened structural coverage sufficiently that a non-macrocycle was predicted to be highly active by Round-03 (bottom of Fig. 4).
Round-00: Initial model building and selectionQuanSA model building begins with an initialization step that produces training molecule alignments. Here, guided by the multiple-ligand alignment shown in Fig. 3, five alternative initial alignments were produced. Having been driven by the same mutual alignment hypothesis, these initial training molecule alignments differed only slightly, but each was used to build a separate QuanSA model. Selection from among alternative models can be done based on statistics derived from the alternative models. These include: (1) model parsimony, which is a quantitative measure of the extent to which molecules with similar activity values have similar predicted poses; (2) Kendall’s Tau for the full re-fitting of training molecules into a derived pocket-field; and (3) the mean unsigned error (MUE) of the re-fit molecules. The alternative quality values are transformed into probabilistic values, and their product reflects the combination of the different metrics. Here, the selected model exhibited a parsimony of 0.63, Kendall’s Tau of 0.87 (CI 0.82–0.91; p \(< 10^\)) and MUE of 0.30 (CI 0.25–0.35).
Figure 5 shows two representative examples from Round-00 for each selection type of candidate molecule. At left (salmon) are the predicted poses for two molecules among the ten predicted most active. As might have been expected, these test molecules have a macrocyclic scaffold in common with the most active training ligand. Also, the right-hand substituents largely occupy the same space as those of UK-2A. Although the activity predictions for compounds Mol-0273 and Mol-0496 were high, these molecules fell within the top 13% and 3%, respectively, of experimental activity within the full future set of 1009.
Fig. 6
Selections of active and informative molecules for Round-01 and Round-02
At right (yellow) are the predicted poses of two molecules predicted to be among the ten most informative candidates. The poses of the test molecules are shown relative to the pose of training molecule UK-2A (green). These four examples are among the twenty molecules selected to refine the current training model. In contrast to the molecules chosen based on high predicted activity, the molecules chosen to be most informative in Round-00 included four non-macrocycles out of the ten chosen (two examples are shown in Fig. 5 at right). Importantly, the predicted 3D alignments compared with that of UK-2A (green) show the new scaffolds in tight congruence to the lower half of the UK-2A macrocycle. Also, the right-hand moieties of the informative molecules had significant surface overlap with those of UK-2A.
Overall, for the 10 predicted to be most active in Round-00, the MUE was quite high (1.7 pIC\(_\) units), but, interestingly, these were all overpredictions. The predicted activity values exceeded even the maximal experimental activity of the most potent training molecule. This characteristic is not typically seen with traditional machine-learning approaches. With most statistical machine-learning methods and deep-learning methods, implicit or explicit modeling of the prior probability of observing a particular prediction value makes out-of-range predictions rare. This is a strength of moving toward a more causal type of predictive model where, for example, the combination of different aspects of multiple active molecules into a new candidate might lead to an out-of-range prediction. Particularly early-on in lead optimization, synthesis of candidate molecules that push the potency envelope is desirable.
Fig. 7
Selections of active and informative molecules for Round-03 and Round-04
Rounds 01-04: Refinement with active learningFigure 6 shows examples of selected molecules for Round-01 and Round-02. Those compounds predicted to be most active retained macrocyclic scaffolds in both rounds, but they showed show some additional diversity in the right-hand hydrophobic groups, with alkyl chains aligning to the benzene moiety of UK-2A (see molecules Mol-0761 and Mol-0415). Also, the the nominal actives were more accurately predicted than for Round-00. For Round-01, the MUE was 1.4 pIC\(_\) units. For Round-02, it was 0.9 pIC\(_\) units, nearly 50% lower than for Round-00, indicating significant refinement in the detailed modeling of a subset of highly active ligands.
Fig. 8
Selections of predicted active candidates for Round-05
Those candidates predicted to be most informative for rounds 01 and 02 contained a higher proportion of non-macrocycles that before. Round-01 had 7/10 non-macrocyclic candidate scaffolds, and Round-02 had 9/10 non-macrocyclic scaffolds (see examples Fig. 6, right, yellow). Alternative branching topologies were seen among the informative candidates as well as novel pendant groups. The flexible thio-ether linkages in compounds Mol-0174 and Mol-0141 still allowed the terminal aromatic rings to overlay well with the corresponding functionality of UK-2A.
One of the challenges in providing computational guidance for synthetic candidate prioritization is having a meaningful explanatory basis for predictions. As shown in Fig. 6, the optimal poses that come out of the fitting process into the quantitative pocket-fields offer convincing correspondence between predictions and known SAR. This is preferable to black-box predictions or those that may yield some explanatory information but do not provide a physically meaningful interpretation.
Figure 7 shows examples of the selected molecules for rounds 03 and 04. By Round-03, when only 60 previous future molecules had been used to refine the original 100-compound model, three non-macrocyclic scaffolds were among the ten predicted to be most active (two examples are shown: molecules Mol-0874 and Mol-0854). A non-macrocycle was also chosen in Round-04 among those predicted most active (Mol-1098, bottom left of Fig. 7). The trend of improvement in accuracy for the predicted most active candidates continued, with an MUE of 0.9 and 0.8 pIC\(_\) units, respectively, for these two refinement rounds.
Those predicted most informative for Round-03 and Round-04 included 3/10 and 5/10 non-macrocycles. The decrease in the number of non-macrocycles in the informative set compared to Round-01 and Round-02 suggests that model refinement improved the predictions on non-macrocycles and thus these molecules would be less represented among those molecules that were being incorrectly predicted as having low activity.
Another aspect of quantitative activity prediction for this series was the clear importance of detailed hydrophobic shape on experimental activity. The ability of a compound to fill the presumed hydrophobic pockets of the non-warhead (right-hand) side of the binding site was a clear activity requirement. Accurately modeling such phenomena depends not only on a molecular representation that captures ligand shape, but also requires that predictions of molecular pose be respectful of the internal conformational energetics of candidate molecules.
Round-05: The goal compoundAs shown in Fig. 8, FPX was chosen by the model that was trained on the initial 100 molecules and subsequently refined with 100 chosen based on the active learning strategy during the ensuing rounds of refinement. FPX was among the 10 predicted to be most active and the activity was accurately predicted with pIC\(_ = 10.0\) with a signed error of just + 0.4 pIC\(_\) units. The MUE of the 10 predicted to be most active was 0.8 pIC\(_\) units, and importantly, the set included 7/10 non-macrocycles, evidence that the model had effectively learned the non-macrocyclic scaffold.
Table 1 Summary of rounds of model building and testingFor FPX, in addition to the predicted pose, a depiction of the quantitative interactions with the pocket-field is shown in Fig. 8. The large majority of interactions were of a purely hydrophobic type, represented by salmon-colored sticks whose length is proportional to the interaction magnitude. Notably, the two fluoro-phenyl groups of FPX, which overlay corresponding hydrophobic functionality of UK-2A, are responsible for significant interactions. In addition, the two chiral methyl groups, especially the one at the lower right, were also important. Because of the angle needed to adequately display the key hydrophobic interactions, the specific polar interactions made by the warhead and the amide linker are somewhat difficult to discern, but all of the specific polar moieties were responsible for key interactions as well (blue and red sticks, for hydrogen-bond donors and acceptors, respectively). The other example highlights both the variability that can be tolerated in the pendant hydrophobic groups and the fact that the core scaffold shifts in accommodating different substituents.
In this gedankenexperiment, only 100 additional future molecules needed to be synthesized, tested, and added to the model in order to correctly choose FPX as an excellent candidate molecule. In completing Round-05, FPX and the other 19 chosen candidate molecules would be synthesized and tested. Overall, only 120/1009 future molecules needed to be “made” to both identify and confirm FPX as a highly active candidate with just two chiral centers, no macrocyclic component, and favorable synthetic characteristics.
Temporal model evolutionTable 1 summarizes the statistics for the rounds of model building, refinement, and future predictions. The training re-fit Kendall’s Tau was consistently high (0.82–0.87) throughout the five rounds of refinement, indicating that model fidelity was maintained as new molecules were added. Likewise, the training re-fit MUE remained low (0.30–0.36 log units) throughout model refinement.
Here, the sets of future molecules were much larger than the training set, and they reflected substantial changes in the structural composition of molecules and the distribution of activity values compared with the training molecules. Later in the project, as expected, a larger proportion of synthesized molecules had very high activity. During successive rounds of scoring future molecules, Tau trended upward, increasing from 0.35 to 0.46. A large proportion of molecules had experimental activity values of 8.5–9.5. The small data range coupled with the presence of assay noise limits the upper bound on rank-based statistics.
More striking was the decrease in MUE for predictions on future molecules from 1.24 to 0.70. The model became significantly more accurate during refinement. As the future MUE decreased, the FPX predicted activity improved from pIC\(_\) = 7.3 (signed error −2.2) in Round-00 to pIC\(_\) = 10.0 (signed error +0.4) in Round-05. Model improvement was further reflected in the predicted rank of FPX activity which rose from the top 61% to the top 1%.
Fig. 9
Experimental versus predicted activities for the set of all future molecules for each round of testing
Table 2 Summary of the distribution of predictions on future moleculesFig. 10
Scheme for generating synthetic ideas using a combination of eSim screening and automatic addition of desirable pendant groups
Figure 9 shows the plots of experimental versus predicted activities for the set of all future molecules for each round of testing. The identity line indicates perfect prediction, and the lighter lines represent \(\pm 1.5\) units of pIC\(_\) (corresponding to \(\pm 2\) kcal/mol). The initial Round-00 model exhibited a strong lower-right triangular bias, with a significantly larger fraction of underpredictions than overpredictions. This aspect of the model’s predictive behavior shifted rapidly with the ensuing two rounds of active learning. By Round-03, relatively little skew was apparent. The distribution of underpredictions (< −2 kcal/mol, Fig. 9, red triangles) decreased 10 percentage points from Round-00 to Round-01 and became nearly as few as the overpredictions in Round-03 to Round-05. The Round-05 FPX prediction (Exp pIC\(_\) = 9.5, Pred pIC\(_\) = 10.0) is highlighted in red.
Table 2 shows a summary of the distribution of predictions on future molecules depicted in the plots of Fig. 9. Throughout model refinement and predictions on future molecules, large overpredictions were few and relatively constant (7% in Round-00 to 3% in Round-05). The predictions within 2 kcal/mol increased from 66% in Round-00 to 91% in Round-05, and those within 1 kcal/mol from 36% in Round-00 to 61% in Round-05. The dramatic decrease in underpredictions occurred in two steps, from Round-00 (27%) to Round-01 (17%) and from Round-02 (14%) to Round-03 (8%). By Round-05, the fractions of large over- and under-predictions were essentially the same.
The distribution of experimental activity values for the future set of molecules changed relatively little over time, perhaps as expected given that only roughly 10% of the molecules were selected over the rounds of iterative refinement. The minimum and maximum pIC\(_\) values were 4.3 and 10.1, respectively, throughout. The mean and standard deviation began with \(8.5\pm 1.0\) and ended with \(8.4\pm 1.0\). However, for the training set, the distribution shifted. The initial minimum and maximum pIC\(_\) training values were 4.3 and 9.5, respectively, shifting to 4.3 and 9.9 at the end of refinement. The mean and standard deviation began with \(7.6\pm 1.3\) and ended with \(8.1\pm 1.2\). The distributional shifts in the training data during refinement reflected the successful selection of numerous potent candidate molecules.
Idea generationOne difficulty in interpreting the results shown in the foregoing is that the set of molecules from which we selected molecules had been made and tested as part of an active design process, where decisions on what to make next were undertaken by experts based on their knowledge of prior data as well as their expertise in the field. So, while the active learning approach was able to efficiently select from that set of molecules, it is not clear that such a path could be followed in a situation where the future space of molecules was open to determination.
Generative approaches for producing ideas for new compounds that employ deep learning have gained some prominence recently [9, 10]. We have taken a different approach, instead using molecular similarity to identify possible bioisosteric core scaffold replacements, including their suitability to display the require pendant functionality for good activity. Figure 10 illustrates how a combination of similarity-based screening and combination with desirable pendant groups can rapidly generate ideas. Our approach is similar in spirit to work by Awale, Hert et al. [23], in which the authors describe a 2D matched-pair approach to identifying sensible candidate molecules based on an analysis of large structure-activity databases.
Beginning with the original five-molecule multiple-ligand alignment used to guide QuanSA model-building, the pendant groups were removed to produce a core-scaffold overlay, and the amide linking subfragment was extracted (Fig. 10A at right). The roughly 3,000,000 compound Enamine Stock Screening collection was screened against the multiple-ligand core using the amide fragment as a required positional restraint to ensure that all hits returned would have appropriate chemistry for linkage to the common warhead. Two examples of high-scoring hits from the screen are shown in Fig. 10B (cyan carbons) in their optimal poses relative to the screening target (green carbons).
For each returned pose of each nominal screening hit, a geometric matching procedure was employed to identify crossover points between the screening hit and each of the full parent molecules from the original multiple-ligand alignment. Figure 10C shows the process using the pose of compound Mol-0013 as the crossover target. When a compatible set of distances and bond vectors existed, the original substituents of the screening hit were replaced with the substituents of the parent compound. Figure 10C (bottom) shows the two resulting merged molecules with novel structures. The arrows and corresponding thick lines show the specific substituent movements that were made. The initial crossover results in high local strain for the new bonds, and the final ligand pose is relaxed using positionally-restrained energy minimization.
Figure 10D shows the relationship of the two resulting generated candidate molecules. Each contains a large fraction of the exact substructure (including chirality) of the final FPX compound, with relatively minor variations in the precise hydrophobic substituents at right. With a slight generalization of the procedure to include additional substituent variations (e.g. p-fluoro-phenyl at both positions), the exact structure of FPX would have been generated. The data required to identify the five-molecule multiple-ligand alignment was just the first 100 compounds from the full structure-activity set. The computational procedure for identifying core-scaffold hits and producing merged candidate molecules required less than an hour and no additional data.
The procedure just described is not intended to fully automate candidate compound generation. Rather, it is meant to be a source of ideas that are easy to scan rapidly. Of course, it is also possible to make use of the predictive QuanSA models to identify candidates that are quantitatively predicted to have high potency or have high information value.
留言 (0)