
記住我
Glucocorticoids are two-faced drugs: they have beneficial anti-inflammatory effects but, on the other hand, trigger complications like osteoporosis; identifying glucocorticoid-mediated pathways responsible for side effects, while maintaining their anti-inflammatory action, has been a long-standing goal. Fu et al. discovered that Tau acts as a low-affinity glucocorticoid receptor to induce bone resorption.
This year marks the 75th anniversary of a pivotal breakthrough in medical science: the successful use of glucocorticoids (GCs) to treat patients with rheumatoid arthritis.1 This achievement earned the biochemists and physicians behind it the Nobel Prize in the following year.1 Since then, GCs have become a cornerstone in managing uncontrolled and aberrant inflammatory responses across a range of conditions.2 Today, between 0.5% and 1% of the population is on chronic steroid therapy.3
However, long-term GC use is not without its downsides. Patients often experience a variety of adverse effects, including increased susceptibility to infections, insulin resistance, weight gain, myopathy, and more. One of the most prominent complications is osteoporosis, with GC-induced osteoporosis (GIO) being the most common form of secondary osteoporosis. Despite considerable efforts to develop bone-sparing GCs or selective GC receptor modulators, success has been limited.1 Research in mice has shown that suppression of bone formation — a hallmark of GIO — is directly mediated by GCs action on osteoblasts via their cognate GC receptor (GR).4,5 In contrast, the mechanisms behind GCs’ long-term effects on bone resorption remain controversial and complex. On the one hand, GCs inhibit osteoclast maturation from myeloid progenitors;6 on the other hand, prolonged treatment has been shown to extend the lifespan and even increase the size of mature osteoclasts.6,7,8 Additionally, indirect effects on osteoblast/osteocyte–osteoclast coupling factors may contribute to the net result of increased bone resorption.9
In a groundbreaking study by Fu and colleagues propose that a receptor other than the GR mediates the effects of GCs on bone-resorbing osteoclasts.10 The surprising receptor for GCs is Tau — a protein previously known primarily for its role in neurodegenerative diseases, such as Alzheimer’s disease.
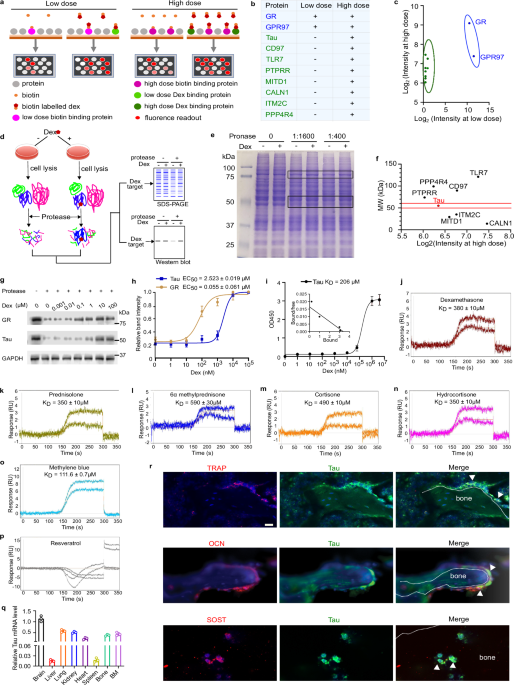
The researchers began their investigation by screening a peptide library of 20,000 proteins derived from ~16,000 coding genes, searching for peptides that bind dexamethasone, a common synthetic GC, with low (400 µM) and high affinities (0.2 µM). Their screen successfully identified the previously known receptors GR and GPR97, but surprisingly, not the mineralocorticoid receptor — a high-affinity GC receptor — suggesting that a number of potential unknown receptors might have been overlooked. Nevertheless, the screen yielded eight proteins that bound dexamethasone at high, but not low concentrations. These were further evaluated using a drug affinity responsive target stability assay. Among these, only Tau exhibited significant protection from protease degradation in this assay. Tau’s involvement in osteoclast regulation was further highlighted through experiments mapping a proline-rich region and identifying the threonine residue at position 212 (Thr212) as critical for its function to mediate dexamethasone effects. While Tau had been primarily associated with brain function, its expression was also detected in bone cells, including osteoclasts, osteoblasts, and osteocytes.
To better understand Tau’s role in GC-mediated bone loss, Fu and colleagues compared mice with an inducible global Nr3c1 (encoding GR) deletion to Tau-knockout animals in a collagen-induced arthritis model. As expected, GR-deficient animals did not respond to the anti-inflammatory effects of GCs. However, Tau-deficient animals exhibited a robust anti-inflammatory response. Notably, trabecular bone loss still occurred in GR-deficient mice, albeit to a lesser extent than in wild-type animals, while Tau-knockout mice were largely protected from bone loss. The suppression of osteoblast function, however, remained GR-dependent (Fig. 1).
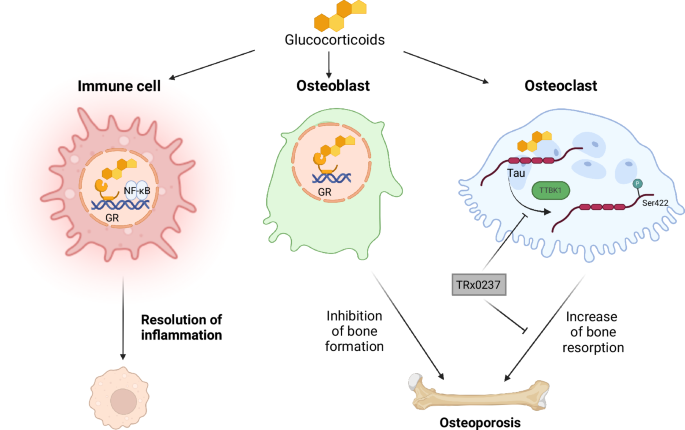
Fig. 1: GCs act on immune and bone cells by different mechanisms.
GCs act through the GR in immune cells to reduce inflammation by interfering with NF-κB activity and other factors. In osteoblasts, they inhibit bone formation. In osteoclasts, GCs bind to Tau and recruit TTBK1, thereby inducing Tau phosphorylation at Ser422, resulting in enhanced osteoclastogenesis and increased bone resorption. The Tau inhibitor TRx023 suppresses Tau action and reduces bone resorption.
In a dexamethasone-induced osteoporosis model, the significant bone loss observed in wild-type mice was reduced in GR-deficient animals and apparently absent in Tau-deficient mice. These findings suggest that while GCs suppress osteoblast function through GR, their effects on osteoclast activity are mediated by Tau. In vitro studies further confirmed that high doses of dexamethasone (10 µM) promoted osteoclastogenesis and resorption in a Tau-dependent, but not GR-dependent, manner. Introducing mutant Tau at Thr212 effectively blocked osteoclastogenesis.
A critical discovery in the study was the identification of Ser422 as another key residue for Tau’s role in osteoclastogenesis. Dexamethasone exposure led to the phosphorylation of Tau at Ser422, a process mediated by Tau tubulin kinase 1 (TTBK1) (Fig. 1). Co-purification experiments revealed that Tau binds to the NF-κB subunit p105, promoting its processing to p50. This p50/p105 NF-κB dimer was activated by high-dose dexamethasone, likely contributing to enhanced osteoclastogenesis.
After elucidating the signaling cascade initiated by dexamethasone, Fu and colleagues sought to identify potential inhibitors of Tau in osteoclast activation. They found that TRx023, an FDA-approved drug that interferes with Tau aggregation, successfully blocked dexamethasone-induced osteoclastogenesis by reducing NF-κB p50 recruitment to the NFATc1 promoter. In a mouse model of GIO, TRx023 treatment partially improved bone quality by impairing osteoclast activity, although suppression of bone formation persisted (Fig. 1). Remarkably, in the context of collagen-induced arthritis, TRx023 restored bone mass and trabecular integrity to normal levels. Elevated Tau phosphorylation observed in dexamethasone-treated mice was also seen in humans with osteoporosis. TRx023 treatment in mice reduced this phosphorylation.
This study represents a remarkable breakthrough, yet several important questions remain. For instance, dexamethasone was used as the model GC; therefore, it is crucial to determine whether Tau also contributes to bone loss mediated by other GCs, such as prednisolone. While the in vitro effects of GCs on osteoclasts via Tau are compelling, the possibility of a broader systemic role cannot be ruled out, as the study utilized global knockout mice. The activation of NF-κB in osteoclasts by GCs through Tau is surprising, particularly since, in other myeloid cells, NF-κB suppression is a hallmark of GC-induced immune suppression.1 This suggests that GC‒Tau interactions may exhibit cell type-specific effects. It would also be intriguing to explore whether tauopathies — characterized by the accumulation of Tau proteins in neurodegenerative diseases — are influenced by GC treatment.
Additionally, the initial screen for low-affinity GC receptors did not encompass the entire human proteome, leaving open the possibility that more low-affinity GC receptors are yet to be discovered, which could play a role in diverse pathological processes. Fu et al. have opened the door for further research into novel GC receptors, potentially leading to more targeted therapies that reduce the side effects of this 75-year-old hormonal drug.
留言 (0)