
記住我
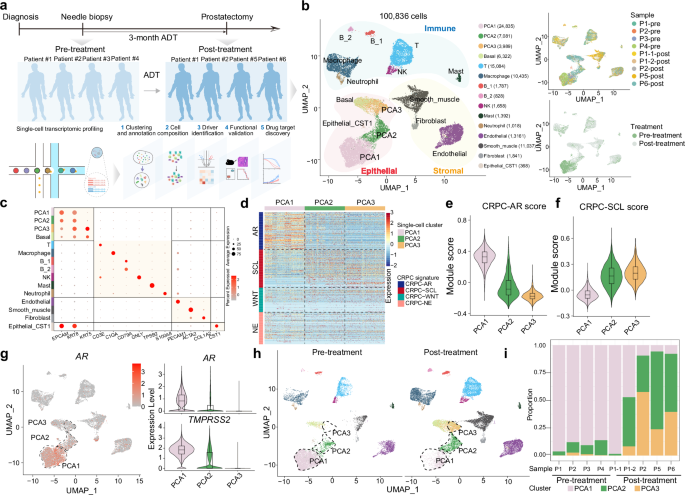
The transition to AR-null lineage, underscored by the rising prevalence post the introduction of AR-targeting therapy like enzalutamide and abiraterone, highlights the role of AR inhibition in driving this change.11 To dissect the impact of ADT on prostate cancer at single-cell level, we collected fresh prostate cancer specimens from patients both before (pre-treatment, via needle biopsy) and after (post-treatment, via radical prostatectomy) 3-month ADT treatment (Fig. 1a; Supplementary information, Table S1). The pathological examination of specimens demonstrated that all of them were prostate cancer. Through single-cell RNA sequencing (scRNA-seq), we analyzed nine specimens from six patients, including paired pre- and post-treatment specimens from two individuals, capturing a comprehensive dataset of 100,836 qualified cells across epithelial, immune and stromal compartments (Fig. 1b, c; Supplementary information, Fig. S1a).
Fig. 1: Single-cell transcriptome profiling of the ADT-induced lineage plasticity.
a Schematic diagram of the experimental design and downstream analysis pipeline to identify the driving forces of lineage plasticity and potential therapeutic strategies in AR-independent CRPC. b Uniform manifold approximation and projection (UMAP) showing the information of 15 single-cell clusters comprising a total of 100,836 cells, the specimens and the treatments. c Dot plot showing the expression levels of the representative markers of each cell lineage. d Heatmap showing the module scores of the four CRPC subtypes, including CRPC-AR, CRPC-SCL, CRPC-WNT and CRPC-NE in PCA1, PCA2 and PCA3. e, f Violin plot showing the module scores of CRPC-AR (e) and CRPC-SCL (f) calculated by the AddModuleScore function implanted in Seurat. g Feature plot showing the expression level of AR (left) and violin plot showing the expression levels of AR and TMPRSS2 (right). h UMAP plot according to the treatment information. i Cell proportions of prostate cancer cells in each of the nine specimens before or after ADT treatment.
We identified three distinct clusters within prostate tumor cells: PCA1, PCA2, and PCA3 (Fig. 1b). Given the classification of CRPC into AR-dependent (CRPC-AR) and neuroendocrine (CRPC-NE) types, alongside two AR-negative/-low subtypes (CRPC-SCL and CRPC-WNT),13 we assessed the identity of these clusters by calculating module scores for each subtype based on signature gene expressions (Fig. 1d). The PCA1 cluster exhibited higher CRPC-AR module score compared to PCA2 and PCA3 clusters (Fig. 1e), suggesting its classification as an AR-positive prostate cancer (ARPC) subtype. Conversely, PCA2 and PCA3 demonstrated significantly higher CRPC-SCL module scores (Fig. 1f), aligning them more closely with the AR-negative/-low CRPC subtype. The module scores for CRPC-NE and CRPC-WNT were minimal across all these three clusters (Supplementary information, Fig. S1b, c). Further analysis of AR expression revealed high levels in PCA1, while PCA2 and PCA3 showed low to negligible expression, consistent with the expression patterns of AR target genes (TMPRSS2, KLK3, and NKX3-1) (Fig. 1g; Supplementary information, Fig. S1d, e). Additionally, the absence of significant ENO2 expression across all these three cell clusters confirmed a lack of neuroendocrine differentiation (Supplementary information, Fig. S1f).
In all five post-treatment specimens, there was a significant decrease in the proportions of total prostate tumor cells among all cell types, indicating the initial clinical efficacy of ADT (Supplementary information, Fig. S1g). Notably, a closer examination of the cellular makeup within the prostate tumor cells revealed that in four out of the five post-treatment specimens, there was an increase in the cell proportions of PCA2 and PCA3 cells, along with a decrease in the proportion of PCA1 cells (Fig. 1h, i). Further trajectory inference analysis suggests that PCA1 could transform into PCA2 and PCA3 (Supplementary information, Fig. S1h–j). Given the CRPC-SCL cell identity and the lack of AR expression in PCA2 and PCA3, these findings suggest that cells within these clusters may exhibit some level of resistance to ADT. Collectively, these observations highlight a shift in the prostate tumor landscape following ADT, moving from AR-positive prostate cancer (PCA1) towards AR-negative/-low prostate cancer (PCA2 and PCA3).
Attenuated LKB1 pathway activity associates with AR independence in human prostate cancerTo explore pathways potentially linked with the transition to AR-null lineage in prostate cancer, we developed a computational pipeline leveraging our scRNA-seq data and the publicly accessible SU2C CRPC cohort14 (Fig. 2a). Firstly, within the SU2C cohort, we categorized 52 samples with high AR expression (AR-high) (top quartile, 52/208) and 52 with low AR expression (AR-low) (bottom quartile, 156/208) (Supplementary information, Fig. S2a, b). As expected, AR target genes such as NKX3-1, FKBP5 and TMPRSS2 were significantly less expressed in the AR-low group (Supplementary information, Fig. S2c–e). Consistent with the basal differentiation observed in some AR-low or -negative CRPCs,13,15 the AR-low group showed increased expression of basal differentiation markers TP63, KRT5, and KRT6B (Supplementary information, Fig. S2f–h). The neuroendocrine phenotype was similarly low across these two groups (Supplementary information, Fig. S2i–k).
Fig. 2: Integration analysis uncovers the attenuated LKB1 pathway activity in AR-independent human prostate cancer.
a Scheme of the computational pipeline to systematically identify the dysregulated signaling pathways in AR-independent prostate cancer. b Heatmap showing the module scores of dysregulated biological pathways across PCA1, PCA2 and PCA3 cells. c Heatmap showing the module scores of dysregulated biological pathways across AR-low and AR-high samples. d Volcano plot showing the 99 upregulated and 69 downregulated pathways based on the scRNA-seq data of this study. e Dot plot showing the activities of LKB1 pathway (REACTOME_ENERGY_DEPENDENT_REGULATION_OF_MTOR_BY_LKB1_AMPK) and AR pathway (REACTOME_ACTIVATED_PKN1_STIMULATES_TRANSCRIPTION_OF_AR_ANDROGEN_RECEPTOR_REGULATED_GENES_KLK2_AND_KLK3) based on the scRNA-seq data of this study. f Volcano plot showing the 99 upregulated and 69 downregulated pathways based on a publicly available bulk RNA-seq cohort, SU2C. g Heatmap showing the activities of LKB1 and AR pathways based on the publicly available bulk RNA-seq cohort, SU2C. h–k Pearson correlation analysis between the expression of AR and the LKB1 pathway component genes, LKB1 (h), CAB39L (i), PRKAG1 (j) or PRKAG2 (k).
Secondly, we assessed the activity of 2227 signaling pathways (including KEGG: 186; Biocarta: 292; Reactome: 1553; and Pid: 196) across individual tumor cells in our scRNA-seq data and tumor samples in the SU2C cohort (Supplementary information, Table S2). This analysis identified 564 pathways upregulated (PCA3 > PCA2 > PCA1, negatively associated with AR expression) and 266 pathways downregulated (PCA3 < PCA2 < PCA1, positively associated with AR expression) from our scRNA-seq data (Fig. 2b). In the SU2C cohort, 290 pathways were upregulated in AR-low samples compared to AR-high samples (AR-low > AR-high), and 410 pathways were downregulated (AR-low < AR-high) (Fig. 2c).
Lastly, integrating these dysregulated pathways from both datasets, we identified 99 upregulated and 69 downregulated pathways showing consistent directional changes across datasets (Fig. 2d, f). Among these pathways, we observed expected dysregulations: AR-related pathways were downregulated, while JAK-STAT pathway16 and FGF-related pathways11 were upregulated (Fig. 2d, f), validating the capability of our computational pipeline to pinpoint dysregulated pathways associated with AR-null lineage plasticity. Notably, LKB1 pathway activity was consistently downregulated in both our scRNA-seq (Fig. 2d, e) and the SU2C cohort (Fig. 2f, g). Further analysis of pathway components revealed a positive correlation between the expression levels of LKB1 (also known as STK11) and AR (Pearson correlation coefficient = 0.28; P = 4.3927 × 10–5) (Fig. 2h; Supplementary information, Fig. S2l). In addition, other LKB1 pathway components, such as CAB39L (Fig. 2i; Supplementary information, Fig. S2m), an activator of LKB1,17 and PRKAG1 (Fig. 2j; Supplementary information, Fig. S2n) and PRKAG2 (Fig. 2k; Supplementary information, Fig. S2o), two downstream effectors, were significantly downregulated in AR-low prostate cancers. Furthermore, the association between LKB1 pathway inactivation and AR independence was consistently validated in two additional publicly available datasets15,18 (Supplementary information, Fig. S2p, q). Collectively, our integrated analysis suggest that attenuated LKB1 pathway activity is linked with AR independence in human prostate cancers.
LKB1 loss promotes prostate cancer progressionTo functionally validate the role of LKB1 pathway in prostate cancer progression, we engineered a genetic model by interbreeding Lkb1flox/flox mice with those carrying Pb-Cre4 and floxed Pten alleles (Pb-Cre4; Ptenflox/flox, PP), which resulted in a prostate epithelium-specific deletion of both Lkb1 and Pten (Pb-Cre4; Ptenflox/flox; Lkb1flox/flox, PPL) (Fig. 3a). We found that the absence of LKB1 remarkably promoted prostate tumor burden (Fig. 3b, c) and enhanced tumor cell proliferation (Fig. 3d). Moreover, LKB1 loss substantially increased the propensity for prostate cancer to metastasize to the lung (88%) and lymph node (92%) in a PTEN-null context (Fig. 3e). In alignment with these findings, the PPL mice exhibited a markedly reduced survival rate in comparison to the PP mice (Fig. 3f). The capacity for organoid formation from freshly isolated PPL cancer cells was significantly higher than that of PP cancer cells (Fig. 3g, h), further confirming the tumorigenic potential induced by LKB1 loss in prostate cancer.
Fig. 3: LKB1 loss promotes prostate cancer progression and metastasis.
a Schematic diagram of construction strategy of Pb-Cre4; Ptenflox/flox; Lkb1flox/flox mouse model. b Gross anatomy of representative prostate tumors of the 15-week-old PPL and PP mice. c Tumor weight quantification of prostate tumors of the 15-week-old PPL and PP mice. d Immunofluorescence staining of DAPI and Ki67 in PP and PPL prostate tumors (left) and the quantification of the percentage of Ki67+ cells in total cells (right). Scale bar, 50 μm. e Gross anatomy of representative metastases to lung and lymph node (top) and quantification (bottom). f Survival statistics of PP and PPL mice. g Cell growth of the organoids derived from freshly dissociated tumor cells of PP and PPL mice. h Organoid formation of freshly dissociated tumor cells of PP and PPL mice. Scale bar, 50 μm.
LKB1 loss promotes prostate cancer lineage plasticity and antiandrogen resistanceSubsequently to evaluating the role of LKB1 inactivation, we delved into its impact on prostate cancer lineage plasticity by comparing lineage marker gene expression between PP and PPL tumors. We noted that accompanying PPL tumor progression, there was a gradual decline in the expression of AR and the luminal marker CK8, alongside an increase in the expression of the basal marker P63 (Fig. 4a). At late stages (12- and 15-week-old), PPL tumors exhibited a mixed lineage, characterized by both AR-low and AR-negative focal differentiation (Supplementary information, Fig. S3a, b). These results suggest that LKB1 inactivation triggers a lineage shift from AR-positive prostate cancer at early stages to AR-negative/-low prostate cancer at late stages.
Fig. 4: LKB1 loss confers AR-independent lineage transition in prostate cancer.
a H&E staining and immunohistochemical staining of LKB1, AR, P63 and CK8 in the prostates of 6-, 9-, 12- and 15-week-old PPL mice and 15-week-old PP mice. b PCA analysis of bulk RNA-seq of the prostate tumors of 15-week-old PP and PPL mice. c Box plot showing the CPM (counts per million) values of Ar, Tmprss2, Krt8, Krt5 and Krt14 in the prostate tumors of 15-week-old PP and PPL mice. d Volcano plot showing the upregulated and downregulated DEGs between PPL and PP tumors. e Heatmap showing the RNA expression of three representative genes for AR, luminal, basal, metastasis and stemness phenotypes. f–h GSEA showing that compared with PP, PPL exhibited significantly lower activity of androgen-AR pathways based on three gene sets, including HALLMARK_ANDROGEN_RESPONSE (f), WANG_RESPONSE_TO_ANDROGEN_UP (g) and NELSON_RESPONSE_TO_ANDROGEN_UP (h). i Survival of PPL mice receiving sham or castration treatment starting from 10 weeks old. j Immunofluorescence staining of DAPI and cleaved caspase-3 in sham- or castration-treated PP and PPL prostate tumors. k Quantification analysis of the ratio of cleaved caspase 3+ cells in total cells in sham- or castration-treated PP and PPL prostate tumors. l Immunofluorescence staining of LKB1, AR, TMPRSS2 and DAPI in LNCaP-derived xenografts with or without LKB1 knockdown. m Immunofluorescence staining of LKB1, AR, TMPRSS2 and DAPI in 22RV1-derived xenografts with or without LKB1 knockdown. Scale bars, 50 μm.
Further analysis of differentially expressed genes (DEGs) was conducted using bulk RNA-seq on the prostate tumors from 15-week-old PPL and PP mice (Supplementary information, Table S3). Principal component analysis (PCA) revealed a distinct transcriptomic divergence between PPL and PP tumors (Fig. 4b). In line with immunohistochemistry findings, LKB1 inactivation significantly downregulated the expression of Ar and its target genes (Fkbp5 and Tmprss2) as well as the luminal marker Krt8 (Fig. 4c–e). Gene set enrichment analyses (GSEA) across three different gene sets uniformly indicated a significant suppression of the androgen pathway in PPL tumors (Fig. 4f–h). The upregulation of multiple lineage plasticity genes, including basal (Trp63, Krt5 and Krt14), metastasis (Snai2, Tgfb1 and L1cam), and stemness (Sox2, Mecom and Sox6) markers was induced by LKB1 loss (Fig. 4c–e), further supporting the enhanced cancer cell state plasticity by LKB1 loss. Neuroendocrine lineage genes such as Syp, Foxa2, and Ascl1 were minimally expressed in both PP and PPL tumors (Supplementary information, Fig. S3c), suggesting a lack of neuroendocrine differentiation. Moreover, we reintroduced LKB1 expression in PPL cancer cells, and demonstrated the restored AR phenotype by LKB1 expression, underscoring the role of LKB1 in regulating lineage plasticity (Supplementary information, Fig. S3d–g).
To further corroborate the lineage plasticity induced by LKB1 inactivation, single-cell transcriptomic profiling was performed on prostate tumors from two 15-week-old PPL mice (Supplementary information, Fig. S4a). Following quality control, a total of 11,374 qualified cells spanning epithelial, stromal, and immune compartments were identified across samples (Supplementary information, Fig. S4b–d). The prostate tumor cells were categorized into three clusters: Tumor_cell_1, Tumor_cell_2 and Tumor_cell_3. An examination of lineage marker expression was conducted to ascertain the cell lineage within these tumor cell clusters. Tumor_cell_1 was found to highly express Ar, whereas Tumor_cell_2 and Tumor_cell_3 exhibited low or negligible Ar expression (Supplementary information, Fig. S4e, f). The expression of Tmprss2 and AR signature scores displayed a corresponding pattern across these clusters (Supplementary information, Fig. S4e, f). Conversely, the basal markers, Krt5 and Trp63, were predominantly expressed in Tumor_cell_2 and Tumor_cell_3 instead of Tumor_cell_1 (Supplementary information, Fig. S4f). Furthermore, analysis of trajectory inference suggested that AR-low/-negative Tumor_cell_2 and Tumor_cell_3 originated from Tumor_cell_1 (Supplementary information, Fig. S4g–i). Thus, these scRNA-seq data validated the lineage plasticity induced by LKB1 loss in prostate cancer.
The lineage transition to an AR-independent state is closely linked with the resistance to AR-targeted therapy. To investigate whether the lineage transition induced by LKB1 inactivation also correlates with a response to castration, we subjected 10-week-old PPL mice to castration and monitored their survival. The results revealed that castration did not improve the survival of PPL mice, confirming the castration resistance imparted by the loss of LKB1 (Fig. 4i). Unlike in PP tumor, where a significant increase in the percentage of cleaved caspase 3+ cells was observed post castration, PPL tumor showed no significant change in the level of cleaved caspase 3+ cells following castration (Fig. 4j, k). These results indicate that LKB1 loss endows prostate cancer with resistance to AR pathway inhibition, aligning with the lineage plasticity induced by LKB1 loss.
Furthermore, to assess the impact of LKB1 inactivation in human prostate cancer, we conducted LKB1 knockdown experiments in two human ARPC cell lines, LNCaP and 22RV1. Consistent with findings from mouse study, knockdown of LKB1 significantly reduced the expression of AR and TMPRSS2 in xenografts derived from both LNCaP and 22RV1 cells (Fig. 4l, m). These results collectively suggest that LKB1 inactivation facilitates an AR-independent lineage transition in both human and mouse prostate cancers.
To further explore the role of LKB1 in the presence of PTEN, we generated the Pb-Cre4; Lkb1flox/flox (PL) mouse model (Supplementary information, Fig. S5a). While LKB1 deletion alone led to prostate cancer with a low incidence, the resulting cancerous regions displayed an AR-low phenotype, characterized by the downregulation of AR (Supplementary information, Fig. S5b, c). This underscores the lineage plasticity regulated solely by LKB1 deletion. Additionally, we crossed Lkb1flox/flox mice with mice carrying both Tmprss2CreERT2 (a luminal cell-specific inducible CreERT2 previously established by our lab19) and floxed Pten (Tmprss2CreERT2/CreERT2; Ptenflox/flox, TP) to initiate cancer specifically in prostate luminal cells following tamoxifen administration (Tmprss2CreERT2/CreERT2; Ptenflox/flox; Lkb1flox/flox, TPL) (Supplementary information, Fig. S5d). Despite the low knockout efficiency of LKB1 in the prostates of TPL mice, compared with the LKB1-intact regions in both TPL and TP prostates, the LKB1-deleted region in TPL prostates distinctly exhibited the AR-low phenotype (Supplementary information, Fig. S5e). These genetically engineered mouse model studies further corroborate that LKB1 inactivation is able to suppress the AR pathway in prostate cancer.
We noticed that the Thr172 phosphorylation level of AMP-activated protein kinase (AMPK) was remarkably downregulated by LKB1 loss (Supplementary information, Fig. S6a). To further clarify whether the effects of LKB1 loss on lineage plasticity is associated with AMPK or not, we introduced exogenous expression of AMPK in PPL cancer cells. We found that AMPK overexpression enhanced AR and luminal phenotype, while attenuated basal phenotype (Supplementary information, Fig. S6b–h). In addition, pharmacological activation of AMPK by the benzimidazole derivative compound 991 (C991) also showed the consistent effects on lineage plasticity (Supplementary information, Fig. S6i–o). Thus, these data demonstrated that the effects of LKB1 loss on lineage plasticity are mediated via AMPK to some degree.
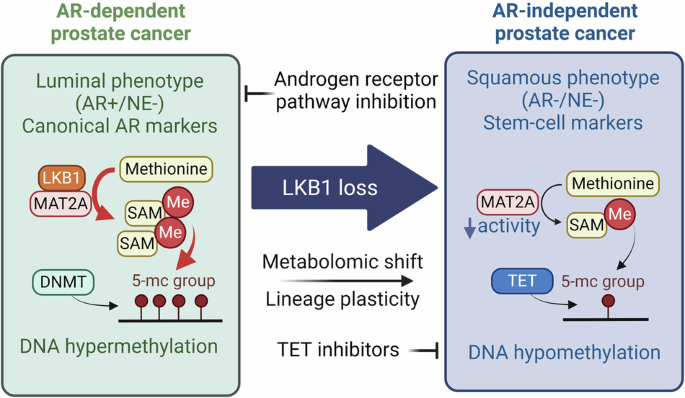
DNA hypomethylation is globally induced by LKB1 lossThe association between lineage transition and epigenetic remodeling is well-documented across diverse biological processes.20,21,22 In the context of LKB1 loss in prostate cancer, an in-depth analysis was conducted to identify the epigenetic factors23 that exhibited differential expression between PPL and PP tumors. This analysis revealed 84 epigenetic factors with increased expression and 41 with decreased expression in PPL tumors compared to PP tumors (Fig. 5a; Supplementary information, Fig. S7a, b and Table S4). Among the downregulated epigenetic factors, Foxa1, which associates with the regulation of prostate cancer lineage plasticity, was identified (Supplementary information, Fig. S7b, c). Notably, among the upregulated epigenetic factors, Tet2 and Tet3 were identified (Fig. 5a), both of which play crucial roles in DNA methylation metabolism, suggesting a potential dysregulation of DNA methylation in PPL tumors.
Fig. 5: Global DNA hypomethylation is induced by LKB1 inactivation.
a Heatmap showing the RNA expression levels of differentially expressed epigenetic factors and box plot showing the CPM values of Tet2 and Tet3 in PPL and PP tumors. b Immunofluorescence staining of 5mC and DAPI in the prostates of 6-, 9-, 12- and 15-week-old PPL mice and 15-week-old PP mice. Scale bar, 50 μm. c PCA analysis of WGBS data of the prostate tumors of 15-week-old PP and PPL mice. d Circos plot globally showing the hypo-methylated and hyper-methylated DMRs across all the chromosomes. Hypo-methylated DMRs are labeled by blue, and hyper-methylated DMRs are labeled by red. e Heatmap showing the methylation levels of hypo-methylated and hyper-methylated DMRs in PPL and PP tumors. f Methylation levels of the hypo-methylated DMRs locating at the promoter regions of Cdk1, Myc, Fgfr2 and E2f7 and the CPM values of these genes.
DNA methylation, a key epigenetic modification in mammals, typically occurs at the fifth carbon of cytosine bases, resulting in 5-methylcytosine (5mC).24 To assess changes in DNA methylation, 5mC levels were examined through immunofluorescence staining, revealing a gradual decrease in DNA methylation correlating with cancer progression in both in vivo tumors (Fig. 5b) and in vitro organoids (Supplementary information, Fig. S7d). For a more detailed analysis of DNA methylation changes, whole-genome bisulfite sequencing (WGBS) was performed on PP and PPL tumors. PCA and clustering analyses highlighted distinct DNA methylation patterns between PP and PPL tumors (Fig. 5c; Supplementary information, Fig. S7e). Subsequent identification of differentially methylated regions (DMRs) between PPL and PP tumors (PPL vs PP) revealed 3250 hyper-methylated and 141,166 hypo-methylated DMRs (Supplementary information, Table S5), indicating a global trend of DNA hypomethylation in PPL tumors (Fig. 5d, e). Given the role of promoter DNA methylation in transcriptional regulation, DMRs within promoter regions (±3 kb around transcription start site) were specifically analyzed and integrated with RNA expression profiles. This analysis showed significant hypo-methylation in the promoter regions of Cdk1, Myc, Fgfr2, and E2f7 along with higher RNA expression levels in PPL tumors compared to PP tumors (Fig. 5f). The oncogenic roles of these genes have been established in multiple cancer types25,26,27,28 suggesting that targeting DNA hypomethylation could be a viable therapeutic strategy in prostate cancer.
Additionally, analyses of differentially methylated loci (DMLs) at single-nucleotide resolution further confirmed the global DNA hypomethylation induced by LKB1 loss (DMLs were listed in Supplementary information, Table S6), with 4914 hyper-methylated and 360,382 hypo-methylated DMLs identified (Supplementary information, Fig. S7f, g). This reinforces the notion that LKB1 inactivation globally promotes DNA hypomethylation, potentially contributing to the observed lineage plasticity and therapeutic resistance in prostate cancer.
To explore the mechanism by which LKB1 loss induces DNA hypomethylation, we determined the levels of SAM and methionine, two key metabolites in DNA methylation metabolic pathway, in both PPL and PP tumors. We found that LKB1 loss significantly downregulated SAM levels, while methionine levels remained unaffected (Supplementary information, Fig. S8a, b), suggesting that the synthesis of SAM from methionine was affected by LKB1 loss. To elucidate how LKB1 regulates SAM levels, we conducted mass spectrometry-based analysis of the LKB1 interactome in PP tumor cells overexpressing Flag-LKB1, and finally identified a total of 176 enriched proteins (Supplementary information, Fig. S8c, d and Table S7). Notably, we discovered and further validated that LKB1 could interact with methionine adenosyltransferase 2A (MAT2A) (Supplementary information, Fig. S8d–g), a rate-limiting enzyme for the synthesis of SAM.29 Moreover, we introduced LKB1 expression in PPL cancer cells, and found that LKB1 expression significantly enhanced MAT2A activity (Supplementary information, Fig. S8h). These data suggest that LKB1 loss attenuates the enzymatic activity of MAT2A, leading to decreased SAM levels and consequent DNA hypomethylation.
Sensitivity of AR-independent prostate cancer to targeting DNA hypomethylationHaving identified the global DNA hypomethylation in AR-independent prostate cancer, we proceeded to investigate the therapeutic potential of counteracting this epigenetic alteration. In mammals, DNA demethylation predominantly occurs through TET-mediated oxidation of 5mC suggesting that inhibiting TET enzymes could promote DNA methylation. We treated PP and PPL organoids in vitro with Bobcat339, a cytosine-based inhibitor of TET.30 Contrary to the negligible impact on PP cancer cells, Bobcat339 remarkably suppressed the proliferation of PPL cancer cells (Fig. 6a, b). This differential sensitivity was underscored by the substantially reduced IC50 (half-maximal inhibitory concentration) value for Bobcat339 in PPL cancer cells (PP: 28.9 μM; PPL: 2.4 μM), indicating a pronounced vulnerability of PPL cancer cells to DNA methylation restoration (Fig. 6c, d). In vivo application of Bobcat339 not only augmented 5mC levels of PPL allograft as expected (Fig. 6e), but also markedly diminished PPL tumor growth (Fig. 6f, g) and significantly extended the survival of PPL mice (Fig. 6h).
Fig. 6: Restoring DNA methylation suppresses AR-independent prostate cancer.
a Organoid formation of freshly dissociated tumor cells from PPL and PP mice under vehicle or Bobcat339 (15 μM) treatment condition. Scale bar, 50 μm. b Quantification of the organoid formation efficiency of PPL and PP cancer cells under vehicle or Bobcat339 treatment condition. c, d Drug sensitivity assay of Bobcat339 in PPL cancer organoids (c) and quantification (d). e Immunofluorescence staining of 5mC and DAPI in the allograft tumors derived from PPL cancer organoids under vehicle or Bobcat339 treatment condition. Scale bar, 50 μm. f Allografts derived from PPL cancer organoids under vehicle or Bobcat339 treatment condition. g Tumor growth of the allografts derived from PPL cancer organoids under vehicle or Bobcat339 treatment condition. h Survival of PPL mice under vehicle or Bobcat339 treatment condition. i Immunofluorescence staining of 5mC and DAPI in the allograft tumors derived from PPL cancer organoids under vehicle or SAM treatment condition. Scale bar, 50 μm. j Organoid formation of freshly dissociated tumor cells from PPL and PP mice under vehicle or SAM (50 μM) treatment condition. Scale bar, 50 μm. k Quantification of the organoid formation efficiency of PPL and PP cancer cells under vehicle or SAM treatment condition. l Schematic diagram showing targeting DNA hypomethylation by exogenous supplementation with SAM and pharmacological inhibition of TETs with Bobcat339.
Furthermore, we explored the effects of augmenting cellular DNA methylation through external supplementation with SAM, a primary methyl donor. SAM administration significantly increased DNA methylation levels (Fig. 6i), and unlike the minimal influence on PP cancer cells, notably inhibited the proliferation of PPL cancer cells (Fig. 6j, k). These results align with the growth-inhibitory effects of TET inhibition by Bobcat339 on PPL cells, rather than PP cells. Given the heightened sensitivity of PPL cells to Bobcat339, these findings reinforce the concept.
Collectively, these data demonstrate the dependency of PPL cancer cell growth on DNA hypomethylation, advocating for the therapeutic strategy of targeting this epigenetic vulnerability through both pharmacological inhibition of TETs and supplementation of SAM to combat AR-independent prostate cancer (Fig. 6l).
Targeting DNA hypomethylation in aggressive prostate cancerFollowing the identification of DNA hypomethylation’s pivotal role in mouse models of prostate cancer, we aimed to extend these observations to human prostate cancer. We assessed DNA methylation status in xenografts derived from multiple human prostate cancer cell lines and organoids using 5mC immunofluorescence staining. Consistent with the findings in AR-independent mouse prostate cancers, AR-negative human prostate cancers exhibited lower levels of DNA methylation compared to AR-positive counterparts (Supplementary information, Fig. S9a). Further investigation into the therapeutic potential of Bobcat339 on a selection of AR-negative/-low prostate cancer organoid lines revealed a significant inhibition of organoid growth under in vitro condition (Fig. 7a–d). Compared to AR-positive cancer cells, AR-negative/-low prostate cancer cells were more sensitive to Bobcat339 treatment (Supplementary information, Fig. S9b, c). Similarly, in vivo studies demonstrated Bobcat339’s capacity to inhibit the growth of MSKPCa1 (Fig. 7e, f) and DU145, an LKB1-negative/AR-negative prostate cancer cell line (Fig. 7g, h).
Fig. 7: Targeting DNA hypomethylation suppresses human AR-independent prostate cancer.
a–d Relative cell viability of AR-negative/-low prostate cancer organoid lines, including MSKPCa1 (a), MSKPCa3 (b), MSKPCa12 (c) and MSKPCa16 (d), under vehicle or Bobcat339 (50 μM) treatment condition. Scale bars, 50 μm. e Immunofluorescence staining of 5mC and DAPI in the xenografts derived from MSKPCa1 organoids under vehicle or Bobcat339 treatment condition. Scale bar, 50 μm. f Tumor growth of the xenografts derived from MSKPCa1 organoids under vehicle or Bobcat339 treatment condition. g Immunofluoresce
留言 (0)