
記住我
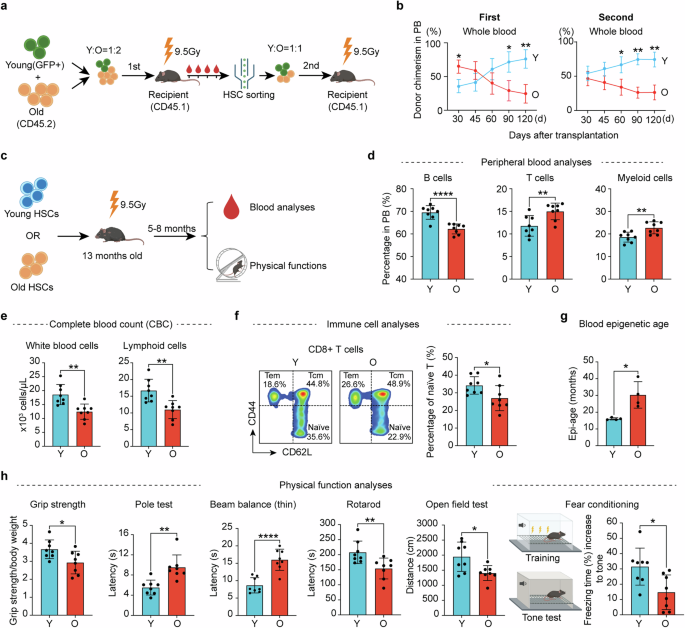
To directly evaluate the functional differences between young and old HSCs in vivo, we sorted long-term (LT)-HSCs (lineage−c-Kit+Sca-1+ (LSK) CD48−CD34−CD150+) (referred to as HSCs) from young and old mice (Supplementary information, Fig. S1a). Consistent with previous reports,12,13,40 a significant increase in the number of HSCs in old mice was observed (Supplementary information, Fig. S1b). Serial transplantation of both young and old HSCs into lethally irradiated young recipient mice (Fig. 1a) revealed a notable decline in the repopulation capacity and biased differentiation of old HSCs compared to their young counterparts (Fig. 1b; Supplementary information, Fig. S1c–f). These results are consistent with previous studies,12,20 and confirm the age-related intrinsic functional decline of HSCs.
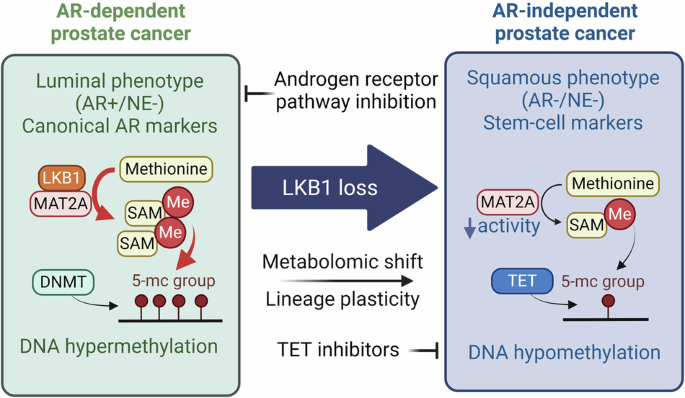
Fig. 1: Transplantation of young HSCs alleviates aging phenotypes in old recipient mice.
a Diagram illustration of the competitive young (2–3 months) and old (22–24 months) HSC transplantation experiment. For the first transplantation, the ratio of young to old HSCs was 1:2 (500 young and 1000 old) while the ratio was 1:1 (1000 young and 1000 old) for the second transplantation. b Peripheral blood (PB) chimerism of donor HSCs at different time points after the 1st and 2nd transplantation, n = 6 for the first and n = 3 for the second transplantation. c Diagram showing the experimental design of individual transplantation of 1000 young (3 months) or 1000 old (22–24 months) HSCs into middle-aged recipients (13-month-old). Five months after the transplantation, a series of hematopoietic and physical tests were performed. d Bar graph showing the percentage of B, T and myeloid cells in the PB of recipient mice, n = 8. e Bar graph showing the absolute number of blood cells in the PB of recipient mice, n = 8. f FACS plot and bar plot showing the percentage of naïve T cells in CD8+ T cells from recipient mice, Naïve T cells (CD44low, CD62Lhigh), Tcm (CD44high, CD62Lhigh) and Tem (CD44high, CD62Llow), n = 8. g Bar plot showing the epigenetic age of blood from recipient mice, n = 4. h Physical tests of recipient mice that received young or old HSCs. Muscle strength, motor coordination, endurance and brain function were assessed, n = 8. Mean ± SD, Student’s t-test, *P < 0.05, **P < 0.01, ****P < 0.0001. The graphics of the mouse and equipment in a, c and h were created with BioRender.
To analyze the contribution of HSCs to systemic aging, we transplanted young or old HSCs into irradiated middle-aged mice (13 months). Five months after transplantation, we evaluated the aging status of the recipient mice by performing blood analyses and physical tests (Fig. 1c). Previous studies have shown that the blood cell composition changes with aging, characterized by an increase in myeloid cells and a corresponding decrease in lymphoid cells.41,42 Notably, mice transplanted with young HSCs exhibited a more youthful blood cell composition, characterized by increased B cell and reduced T cell and myeloid cell proportions, as well as an increase in overall white blood cells and lymphoid counts, compared to the recipients that received old HSCs (Fig. 1d, e). This suggests a crucial role of HSCs in systematic hematopoietic aging.
The ratio of naïve T cells serves as a critical indicator of immune function, which declines with aging.43 Conversely, central memory T cells (Tcm) and effector memory T cells (Tem) exhibit an increase with aging,44 which were confirmed by our study (Supplementary information, Fig. S2). Notably, the old mice that received young HSCs have more naïve T cells in both CD4 and CD8 T cells (Fig. 1f; Supplementary information, Fig. S3a, b), indicating an improved immune system after receiving young HSCs transplantation.
The DNA methylation clock is widely used as an indicator of aging.45 Importantly, old mice that received young HSCs have a younger epigenetic age in blood compared to those that received old HSCs (Fig. 1g). Consistently, old mice that received young HSCs exhibited improved physical functions compared to those that received aged HSCs, including muscle strength, motor coordination, locomotor activity and cognitive functions (Fig. 1h), without significant difference in body weight and spatial memory (Supplementary information, Fig. S3c–e). Collectively, these results demonstrate that transplantation of young HSCs to old mice can alleviate aging-related phenotypes.
scRNA-seq reveals a “younger” HSC subpopulation in old miceThe above results suggest that HSCs from old mice are functionally defective compared to those from young mice. To understand the defects, we performed both bulk RNA-seq and scRNA-seq on young and old HSCs (Fig. 2a), which revealed a clear transcriptomic difference between young and old HSCs (Fig. 2b; Supplementary information, Fig. S4a). Bulk RNA-seq revealed 332 upregulated genes in old HSCs, including previously annotated HSC aging marker genes,46 including Clu, Selp, Mt1 and Ramp2 (Supplementary information, Fig. S4a and Table S1). These genes were defined as HSC aging marker genes for downstream analysis. Interestingly, unsupervised clustering analysis of scRNA-seq revealed that while the quiescent young HSCs are largely uniform, the quiescent old HSCs can be further divided into 3 clusters (q1–q3) (Fig. 2c, d). This result indicates that quiescent old HSCs are transcriptionally more heterogeneous compared to young HSCs.
Fig. 2: scRNA-seq reveals increased heterogeneity of old HSCs.
a Workflow of 10X scRNA-seq of young and old HSCs. The HSCs were sorted from young (2–3 months) and old (23 months) mice. b UMAP plot showing the distribution of young and old HSCs based on scRNA-seq. Clear separation of young and old HSCs indicates transcriptional changes of HSCs during aging. c Cell cycle phase analysis of young and old HSCs based on scRNA-seq. The S-phase and G2/M-phase marker genes are from Seurat package (V4.0.2). The cell cycle phase is determined by the relative expression levels of these marker genes. If neither S-phase nor G2/M phase genes are expressed, they are classified as G0/G1 phase. d UMAP plot showing unsupervised clustering of young and old HSCs. In total, 6 clusters were identified with two clusters (a1 and a2) representing active, and four clusters (q1–q4) representing quiescent cells. e Heatmap showing the cluster-specific marker genes expression across the 6 clusters and their enriched GO terms. Differentially expressed genes with min.pct = 0.25, logfc.threshold = 0.25 among the 6 clusters were used to generate the heatmap. Well-known HSC aging-related genes in clusters q1 and q2 are highlighted. Commonly identified marker genes in clusters q3 and q4 were also highlighted. Exp., expression; Reg., regulation. f Box plot showing relative expression of marker genes of q1, q2 and q3 in young and old HSCs via analysis of bulk RNA-seq. The expression level was normalized to the average expression level of the old. Plot shows the mean and 5–95 percentile. Two-sided unpaired Wilcoxon test. g UMAP presentation of the well-known HSC aging marker genes, Sbspon, Gpr183, Clu and Ramp2 in each of the single cells. h UMAP plot and violin plot showing the calculated aging score of single cells of different clusters based on the HSC aging genes identified in bulk RNA-seq. In total, 332 upregulated genes in old HSCs were used for aging score calculation. Two-sided unpaired Wilcoxon test. **P < 0.01, ****P < 0.0001.
We then identified the marker genes for different clusters (Fig. 2e; Supplementary information, Table S2). Intriguingly, although q1–q3 are all from old HSCs, the known aging marker genes, such as Mt1, Nupr1, Cavin2, Clu, Ramp2, Alcam, Selp and Gpr183, are highly expressed in q1 and q2 clusters, but not in q3 cluster (Fig. 2e). In contrast, q3 and q4 (young HSCs) clusters share highly expressed genes, despite the q3 cluster originating from old mice. Gene Ontology (GO) analysis of q3 marker genes revealed an enrichment of cell proliferation-related pathways (Fig. 2e), which were also enriched in the young HSCs (Supplementary information, Fig. S4b, aging-down). We further calculated the relative expression levels of q1, q2, and q3 marker genes in old and young HSCs, and found that the q1 and q2 marker genes are expressed significantly higher in old HSCs, while q3 marker genes are higher in young HSCs (Fig. 2f), supporting a younger transcriptome of q3 HSCs.
To gain further support for the notion that a subset of HSCs from old mice are “younger” in their transcriptome, we analyzed the UMAP distribution of some well-known HSC aging marker genes (Sbspon, Gpr183, Clu, Ramp2)46 and found that they are expressed at a higher level in q1 and q2 compared to that in q3 (Fig. 2g). In contrast, the young HSC marker genes, such as Rnase6 and Arhgap30, exhibited an opposite expression pattern (Supplementary information, Fig. S4c). To quantify the aging heterogeneity of old HSCs, we calculated the aging score using the 332 HSC aging marker genes (Supplementary information, Table S1) or the top 100 reported HSCs aging genes (Supplementary information, Table S3).46 Both analyses indicate that cluster q3 has a lower aging score than q1 and q2, despite all cells originating from the same old mice (Fig. 2h; Supplementary information, Fig. S4d). Collectively, comparative scRNA-seq analysis revealed increased aging heterogeneity in old HSCs and a subset of old HSCs that have a younger transcriptome.
CD150 can serve as an aging heterogeneity marker for old HSCsTo better understand the aging heterogeneity of old HSCs and to assess its contribution to overall body aging, a unique cell surface marker that facilitates the separation of “younger” from “older” cells in aged HSCs is needed. To this end, we first identified the top 150 genes showing strong correlation with aging scores in scRNA-seq (Supplementary information, Fig. S5a) and compared them with the 332 genes upregulated in old HSCs (Supplementary information, Fig. S4a), resulting in 54 shared genes (Fig. 3a; Supplementary information, Table S4). We further required membrane location for sorting purposes, which reduced the candidate gene list to 26 (Fig. 3a). Furthermore, the potential marker genes should have lower expression levels in q3 than in q1 and q2, which further narrowed the list to 7 (Supplementary information, Fig. S5b). Considering the availability and specificity of the antibodies, CD150 (Slamf1) was selected as the marker for aging heterogeneity of old HSCs. FACS analysis of HSCs from young and old mice indicated that the population of HSCs with higher CD150 levels significantly increases with aging (Fig. 3b), supporting the use of CD150 as an indicator of aging heterogeneity in old HSCs.
Fig. 3: Identification of CD150 as an HSC aging heterogeneity marker.
a Workflow for identifying heterogeneity marker genes in old HSCs. b FACS plot showing expression level of CD150 in young (3 months) and old (22–24 months) HSCs. c HSCs from old mice were separated into four subgroups (25% for each) based on their CD150 protein levels and subjected to bulk RNA-seq, n = 3. d Dot plot showing Pearson correlation between CD150 signature score and aging score based on scRNA-seq data, R = 0.78. e Heatmap showing changes in expression of aging-related genes with ascending level of CD150 based on bulk RNA-seq in c. f Bar plot showing the epigenetic age of CD150low and CD150high HSCs from 22- to 24-month-old mice, n = 4, paired t-test. The HSC subsets from the same mice were paired with dashed line. g Line plot and heatmap showing ATAC-seq signal difference between CD150low and CD150high HSCs in aging-related open and closed regions, respectively. h Diagram of competitive transplantation to evaluate the repopulation capacity of CD150low and CD150high HSCs from old mice. i Whole blood chimerism of CD150low and CD150high HSCs from 22- to 24-month-old donor mice at different time points after the 1st and 2nd transplantation, n = 6 for the first and n = 3 for the second transplantation. Mean ± SD, Student’s t-test, *P < 0.05, **P < 0.01, ****P < 0.0001. The graphic of the mouse in h was created with BioRender.
To further confirm that the CD150 level can serve as a marker of aging heterogeneity of old HSCs, we separated old HSCs into 4 groups (G1 to G4) by FACS based on CD150 levels and profiled their transcriptomes (Fig. 3c). We then calculated the expression correlation between CD150 and each gene and identified 131 and 103 genes that exhibit strong positive or negative correlation with CD150 in aged HSCs, respectively (Supplementary information, Fig. S5c and Table S5). Notably, the genes that positively correlate with CD150 include many known HSC aging markers, including Sbspon, Ehd3, Clu, Selp, Jam2 and Enpp5. In contrast, the negatively correlated genes include genes that are highly expressed in young HSCs, such as Serpinb1a, Usp6nl, Lrr1 and Kif15 (Supplementary information, Fig. S5d). These results suggest that CD150 level can be a potential indicator of HSC transcriptome age.
Using the 131 genes that positively correlate with the CD150 level (referred to as CD150 feature genes), we calculated the CD150 feature score and found that q3 HSCs have a significantly lower CD150 feature score than q1 and q2 HSCs (Supplementary information, Fig. S5e, f). Notably, a strong correlation between the CD150 feature score and aging score was observed (Fig. 3d). This indicates that the aging heterogeneity of old HSCs can be well reflected by CD150 feature genes. Consistently, HSC aging-related up- and down-regulated genes in the 4 groups of HSCs from G1 to G4 exhibit a trend that is similar to the aging process (Fig. 3e), confirming the positive correlation between CD150 level and HSC aging status in old mice.
CD150low HSCs from old mice have younger epigenome and superior functionsEpigenetic changes are believed to be one of the drivers of aging.47,48,49 To further determine whether CD150 level can reflect epigenetic aging in old HSCs, we first compared their epigenetic age and found that CD150low HSCs have a lower epigenetic age than that of CD150high HSCs despite that they are from the same old mice (Fig. 3f). Given that stem cell aging is accompanied by chromatin accessibility changes,50,51,52 we compared the chromatin accessibility landscapes of old CD150low and CD150high HSCs, as well as young and old HSCs by performing ATAC-seq analyses. We found that 4694 peaks displayed increased chromatin accessibility and 3842 peaks showed decreased accessibility in aged HSCs relative to young HSCs, while 3066 open and 1966 closed differential peaks were identified between old CD150low and CD150high HSCs (Supplementary information, Fig. S5g, h). Interestingly, a similar change between CD150low and CD150high old HSCs was also observed around the aging-related ATAC-seq peaks (Fig. 3g). These results indicate that CD150 can serve as a marker reflecting both transcriptional and epigenetic aging in aged HSCs.
When we further checked the genes close to the differential accessible peaks, we found several HSC aging-related genes, including Clu, Jam2, Mt2, Nupr1, Selp, Slamf1 (CD150) and Vwf, are located near the aging-related open peaks, and these genes also harbor the open peaks when comparing old CD150high and CD150low HSCs (Supplementary information, Fig. S5g, h). Consistently, the genes located around closed differential accessible peaks in both comparisons include many highly expressed genes in young HSCs, such as Cd52, Cdc6, Haao and Nkg7, demonstrating the corresponding chromatin accessibility changes underlying transcriptome alteration.
In addition, GO analysis of the genes related to the differential accessible peaks revealed enrichment of ‘cell adhesion’ and ‘potassium transport’ in the open peaks in both comparisons (Supplementary information, Fig. S5g, h). Previous studies have demonstrated a correlation between ‘cell adhesion’ with HSCs functions and aging,46,53,54 further supporting the potential functional defects in old and CD150high HSCs compared to their counterparts.
To gain further insight into the aging-related open and closed ATAC-seq peaks, we performed motif enrichment analysis. We found that the aging-related open peaks are enriched for binding sites of the AP-1 family transcription factors (TFs), while the aging-related closed peaks are enriched for the E26 transformation-specific (ETS) family TFs, including SPIB and PU.1 (Supplementary information, Fig. S5i). Importantly, similar TFs were identified when comparing CD150low and CD150high old HSCs (Supplementary information, Fig. S5j), further supporting a relatively younger chromatin in old CD150low HSCs compared to the CD150high HSCs. AP-1 family TFs have been reported as pioneer factors in aging,55,56 while PU.1 and SPIB play important roles in regulating hematopoiesis,57,58 further highlighting the altered chromatin status and potentially impaired functions in the old CD150high HSCs.
Previous studies have shown that increased DNA damage and decreased percentage of cells in proliferation are associated with functional decline of old HSCs.27,59,60 Interestingly, we observed an increase of double stranded breaks (indicated by γH2AX) as well as a decrease in the proportion of cells in proliferation in CD150high compared to CD150low old HSCs (Supplementary information, Fig. S5k, l), indicating that CD150low HSCs may have better functions than CD150high HSCs in old mice.
To directly compare the functions between old CD150low and CD150high HSCs, we performed serial competitive transplantation (Fig. 3h). We found that CD150low old HSCs have significantly better engraftment capacity compared to their CD150high counterparts in both the first and second transplantation (Fig. 3i; Supplementary information, Fig. S6a, b). These results demonstrate that CD150 level not only marks transcriptome and epigenome heterogeneity, but also reflects functional heterogeneity of old HSCs.
Despite old CD150low HSCs being molecularly and functionally younger than CD150high HSCs, the extent to which they are similar to young HSCs remains unclear. To this end, we conducted molecular and functional comparisons between young HSCs and old CD150low HSCs. The transcriptome comparison showed that, although old CD150low HSCs are younger than CD150high HSCs, they are still different from young HSCs in the expression of aging-related genes (Supplementary information, Fig. S6c). In terms of chromatin accessibility, old CD150low HSCs are similar to young HSCs in aging-related closed regions but have higher signals in aging-related open regions (Supplementary information, Fig. S6d). These results demonstrate that old CD150low HSCs are younger than old CD150high HSCs, but still older than young HSCs at the molecular level. As for the direct functional comparison, based on the competitive transplantation experiment (Fig. 3h), we found that old CD150low HSCs retain about 72.9% functionality relative to that of the young HSCs in terms of their repopulation capability, while the old CD150high HSCs only retain about 5.9% (Supplementary information, Fig. S6e). Collectively, these results indicate that although old CD150low HSCs are not as youthful as young HSCs at the molecular and functional levels, they retain most of the functionality of young HSCs.
Although young HSCs exhibit a homogeneous transcriptome (Fig. 2d) and aging score (Fig. 2h), their CD150 levels are variable (Fig. 3b). This raised the question whether CD150low HSCs are also functionally superior to CD150high HSCs in young mice. To address this question, we checked the cell cycle and performed similar HSC transplantation experiments using young HSCs (Supplementary information, Fig. S7a, b). Consistent with their transcriptome and aging score homogeneity, no significant functional difference was observed between CD150low and CD150high young HSCs for cell cycle analysis or peripheral blood (PB) chimerism during the period of 120 days after transplantation (Supplementary information, Fig. S7a–d). However, we noted a myeloid bias and significantly higher contribution to myeloid cells in young CD150high HSCs, along with an upward trend in PB chimerism when compared to CD150low young HSCs (Supplementary information, Fig. S7c, d). These results indicate that CD150high HSCs from young mice have comparable or even better functions compared to CD150low HSCs, which is in contrast to those in old mice. Furthermore, these findings also suggest that myeloid-biased differentiation is not linked to aging status or decreased functionality in young HSCs.
Old CD150high HSCs are defective in differentiation, but not in self-renewal or activationWe next attempted to dissect the mechanism underlying the differential repopulation capacity of CD150low and CD150high HSCs in old mice. Considering that self-renewal and differentiation are the two major features of stem cells, we compared both features after their transplantation. We first determined the differentiation dynamics of old HSCs at different time points after transplantation (Supplementary information, Fig. S8a). We found that transplanted old HSCs start to differentiate on day 7, and the progenitor subpopulation becomes apparent on day 10 (Supplementary information, Fig. S8b). Based on this, we separately transplanted old CD150low and CD150high HSCs and analyzed the donor-derived hematopoietic stem and progenitor cells (HSPCs) in bone marrow on days 7 and 14 after transplantation (Fig. 4a). Notably, we found that CD150high HSCs showed a significantly lower ratio in short term HSCs (ST-HSCs) and multipotent progenitors (MPPs), but a higher ratio in LT-HSCs compared to CD150low HSCs (Fig. 4b; Supplementary information, Fig. S8c), indicating that old CD150high HSCs might have differentiation and/or activation defects.
Fig. 4: Differentiation, but not self-renewal, is a major defect of old CD150high HSCs.
a Diagram illustration of the transplantation experiment comparing old CD150low and CD150high HSCs. Donor HSCs-derived HSPCs from the bone marrow were analyzed on days 7 and 14 after transplantation. b Representative FACS analysis of donor HSCs-derived HSPCs (left) and quantification of different cell populations 7 days after transplantation (right). LT-HSC (CD45.2 LSK, CD34−CD48−CD150+), ST-HSC (CD45.2 LSK, CD34+/CD48+CD150+) and MPPs (CD45.2 LSK, CD34+/CD48+CD150−) were analyzed, n = 3. c Diagram illustration of the competitive transplantation for evaluating the long-term differentiation of CD150high HSCs. Both PB and bone marrow were analyzed five months after transplantation. d Representative FACS analysis of donor HSC-derived HSPCs (left) and quantification of different cell populations (right) 5 months after transplantation, n = 3. e Bar graph showing the chimerism of donor HSCs in LT-HSCs, ST-HSCs, MPPs and PB 5 months after transplantation, n = 3. f UMAP plot showing cell distribution of HSPCs derived from CD150low and CD150high HSCs in scRNA-seq 14 days after transplantation. g UMAP presentation of cell types predicted based on HSPC marker gene expression (left). In total, 7 different cell types were identified in HSPCs. h Bar graph showing relative abundance of predicted cell types in HSPCs from mice that received old CD150low or CD150high HSCs. Mean ± SD, Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001. The graphics of the mouse in a and c were created with BioRender.
To distinguish between these two possibilities, we profiled transcriptomes of CD150low and CD150high HSCs 4 days after transplantation when the HSCs had yet to initiate differentiation (Supplementary information, Fig. S8d). By comparing with freshly isolated HSCs, we identified 794 commonly upregulated genes in both CD150low and CD150high HSCs after transplantation (Supplementary information, Fig. S8e and Table S6). The commonly upregulated genes are highly enriched in terms related to cellular division. Consistently, the G2M checkpoint-related genes were similarly upregulated in both CD150low and CD150high HSCs upon transplantation (Supplementary information, Fig. S8f). These results indicate that old CD150high HSCs can be effectively activated as that of CD150low HSCs following transplantation.
We further compared the proliferation capacity of old CD150low and CD150high HSCs in culture and found that old CD150low and CD150high HSCs exhibited comparable proliferation rates (Supplementary information, Fig. S8g–k). Taken together, these results indicate that impaired repopulation capacity of old CD150high HSCs is caused by defective differentiation, but not activation or proliferation.
To rule out a possible delay in differentiation of old CD150high HSCs after transplantation, we analyzed the persistence of the differentiation and self-renewal capacity of old CD150high HSCs over an extended period of 5 months. We co-transplanted old CD150low or CD150high HSCs (GFP–) with competitors (GFP+) and analyzed the differentiation and chimerism of donor HSCs 5 months later (Fig. 4c). We observed a consistent differentiation defect of old CD150high HSCs when compared to their CD150low counterparts by analyzing donor-derived HSPCs in the bone marrow (Fig. 4d). Additionally, the donor chimerism of old CD150high HSCs exhibits a clear trend of decline from LT-HSCs through ST-HSCs, MPPs, to PB, while the donor chimerism of old CD150low HSCs remains relatively stable (Fig. 4e), supporting a long-term differentiation defect of old CD150high HSCs.
Collectively, these results indicate a long-lasting impairment in differentiation, but not in activation or self-renewal of old CD150high HSCs.
Old CD150high HSCs are defective in the LT-HSCs to ST-HSCs transitionTo determine the specific stage when the differentiation defects of old CD150high HSCs occur, we performed comparative scRNA-seq analysis of the HSPCs derived from old CD150low and CD150high HSCs 14 days after transplantation. We obtained 2406 and 1925 high quality HSPCs derived from transplanted CD150low and CD150high HSCs, respectively (Fig. 4f). Cell cycle phase analysis indicated that similar proportions of HSPCs derived from CD150low and CD150high HSCs were actively cycling (Supplementary information, Fig. S9a), consistent with our previous results (Supplementary information, Fig. S8e, f).
Based on the expression of the HSPC marker genes50,61 and the HSPC sorting markers (Ly6a, Kit, Cd34, Cd48 and Slamf1), the 4331 HSPCs can be divided into 7 subclusters that include LT-HSC-like, ST-HSC-like, MPP2-like, MPP3-like, MPP4-like, GMP-like and MEP-like cells (Fig. 4g; Supplementary information, Fig. S9b). When HSPCs derived from CD150low and CD150high HSCs were compared, we found that HSPCs derived from CD150high HSCs were significantly enriched in LT-HSC-like cells (Fig. 4g, h), consistent with our FACS analysis (Fig. 4b; Supplementary information, Fig. S8c). Since the proportions of CD150low and CD150high HSCs-derived ST-HSCs are comparable, our data indicate that the differentiation defects of CD150high HSCs were mainly at the stages of LT-HSC to ST-HSC, and ST-HSC to MPP4 transitions. Given that MPP4 are the major progenitor cells of the lymphoid lineage, the substantial decrease in the MPP4 population might be a major cause of the biased differentiation of CD150high HSCs.30,32
Consistently, when we further performed pseudo time analysis to map the differentiation trajectory, we found that HSPCs derived from CD150high HSCs were predominantly enriched at early stages and less enriched at later stages compared to HSPCs derived from old CD150low HSCs (Supplementary information, Fig. S9c), supporting that the differentiation of CD150high HSCs is impaired compared to their CD150low counterparts.
To further analyze the transcriptome differences between populations derived from old CD150low and CD150high HSCs, we identified differentially expressed genes between them. Significant transcriptome differences were observed in LT-HSC-like, ST-HSC-like, and MPP4-like subpopulations (Supplementary information, Fig. S9d–f). Interestingly, some HSC aging marker genes, such as Vwf, Nupr1, Ifitm3, and Ifitm1 are consistently upregulated in HSPCs derived from old CD150high HSCs, indicating that upregulation of HSC aging genes can persist in downstream cells.
To further investigate the transcriptome difference, we performed GO analysis. Upregulated genes in cells derived from CD150low HSCs are consistently enriched for ‘translation-related pathways’ across cell types, while those from CD150high HSCs are enriched for ‘protein folding response-related pathways’ (Supplementary information, Fig. S9g). Increased translation in HSCs is associated with differentiation activities,62,63,
留言 (0)