
記住我
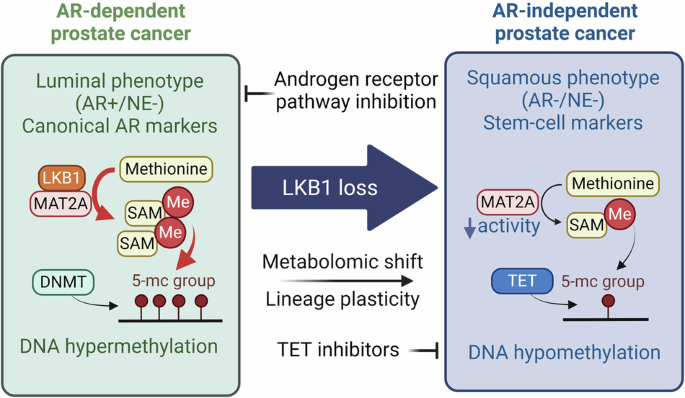
While it is well established that androgen receptor pathway inhibition leads to the development of double-negative prostate cancer (DNPC), the mechanisms underlying this cell fate decision remain unclear. In a recent study published in Cell Research, Li et al. identified that LKB1 inactivation supports the development of DNPC through metabolic and epigenetic rewiring, highlighting DNA hypomethylation as a vulnerability and TET2/3 as potential therapeutic targets.
Lineage plasticity contributes to treatment resistance by establishing an alternative identity, as seen in castration-resistant prostate cancer (CRPC) treated with androgen receptor (AR) pathway inhibitors (ARPIs). In a subset of patients, an AR-independent phenotype emerges, with a subtype characterized by loss of AR and expression of squamous markers rather than neuronal markers, known as double-negative prostate cancer (DNPC).1 DNPC is clinically and molecularly distinct from other subtypes and is associated with the worst survival outcomes among metastatic CRPC patients treated with ARPIs.2
DNPC features loss of TP53, RB1 and PTEN.2 It has distinct chromatin accessibility3 and DNA methylation2 patterns, highlighting the importance of epigenetic modification underlying its progression. Specifically, DNPC is characterized by elevated KRT7 expression and progenitor-like cells expressing SOX2 and FOXA2.4 Although the drivers of DNPC are not fully understood, key factors have been identified, including the amplification of chromatin remodeler CHD7, increased KLF52 and FGF/MAPK activity,5 as well as co-activation of HGF/MET and Wnt/β-catenin.6 While it is established that ARPIs induce epigenetic alterations7 leading to DNPC,2,3,6 the mechanism underlying cell fate decision-making in this context remains incompletely defined (Fig. 1).
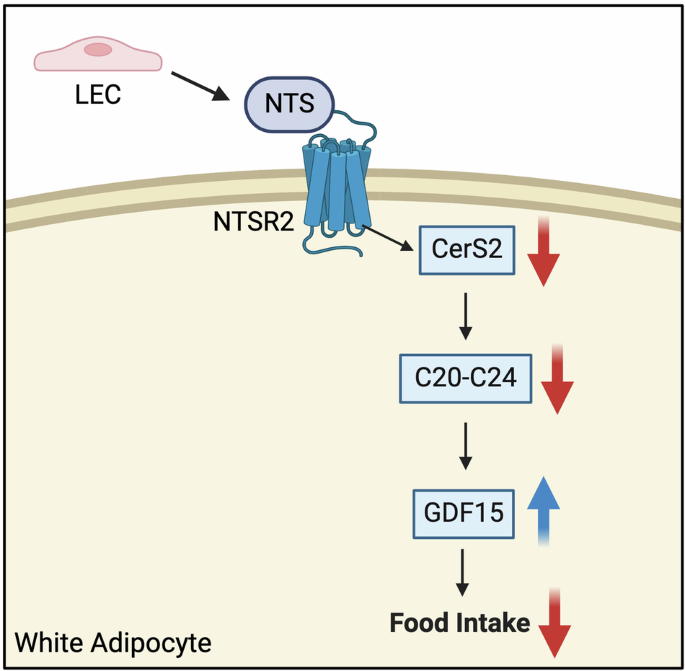
Fig. 1: LKB1 inactivation promotes lineage plasticity and the development of DNPC under AR pathway inhibition.
In AR-dependent prostate cancer, LKB1 binds to MAT2A, enhancing its activity. However, LKB1 inactivation following AR pathway inhibition, leads to the induction of lineage plasticity and a metabolic and epigenetic shift to an AR-independent phenotype.
In this issue of Cell Research, Li et al.8 delve into the molecular mechanisms contributing to the development of lineage plasticity in response to androgen deprivation therapy (ADT) and progression to DNPC. Using single-cell transcriptomic profiling of patient needle biopsies pre- and post-ADT, they identified clusters including AR-positive prostate cancer (ARPC) and AR-negative/low CRPC subtypes enriched with stem-like programs. They noted a shift in cell populations from ARPC towards AR-negative/low in response to ADT. A subsequent gene set enrichment analysis of genes differentially expressed between ARPC and AR-null lineage in single-cell RNA-seq as well as in SU2C CRPC cohort, identified downregulation of the LKB1 pathway associated with AR-null lineage.
LKB1 is a serine-threonine liver kinase encoded by STK11. LKB1 is a master kinase that directly phosphorylates and activates AMP-activated protein kinase (AMPK) and suppresses mTOR signaling. LKB1 functions as a tumor suppressor and has key roles in cell growth, metabolism and polarity.9 While alterations in LKB1 are common across various cancers, homozygous deletions and point mutations are rare in ARPC. However, in metastatic prostate cancer, the frequency of LKB1 genomic aberrations increases.10 Beyond its role as a metabolic checkpoint regulating AMPK/mTOR signaling, the mechanisms by which LKB1 inactivation unleashes lineage plasticity and drives prostate cancer progression remain unclear.
Using genetically engineered mouse models, the authors found that LKB1 loss remarkably promotes prostate tumor burden, metastasis, and reduces survival. Transcriptional profiling of LKB1-inactivated tumors identified suppression of AR and luminal phenotype, increased basal and stemness markers, and activation of mixed lineage populations. Notably, the LKB1 knockout tumors were resistant to castration. These observations were consistent with the shift from ARPC to an AR-independent phenotype associated with resistance to ADT, which is enriched with stem-like, basal, or neuroendocrine traits. Single-cell transcriptomic profiling of mouse tumors corroborates the induced lineage plasticity imparted by LKB1 inactivation. Altogether, the authors provided a clear association between LKB1 inactivation and induction of lineage plasticity, particularly the transition from ARPC state to the early stages of AR-negative/low in human and mouse prostate cancer.
An in-depth analysis to uncover epigenetic factors contributing to lineage plasticity and the AR-null phenotype following LKB1 inactivation led to the identification of TET2 and TET3. The TET family is known for its role in regulating DNA methylation, where they catalyze the sequential oxidation of 5-methylcytosine to 5-hydroxymethylcytosine (5hmC) and ultimately lead to the demethylation of cytosine. 5hmc has been implicated in delineating lineage plasticity and transdifferentiation,11 with the TET family playing a crucial role in lineage specification. Using whole-genome bisulfite sequencing, they explored DNA methylome following LKB1 inactivation and revealed a distinct DNA methylation pattern in LKB1-inactivated tumors. Specifically, a global trend of DNA hypomethylation was associated with LKB1 inactivation. Mechanistically, they showed that in ARPC, LKB1 forms a complex with MAT2A, the enzyme responsible for synthesizing methionine to S-adenosylmethionine (SAM), the global methyl donor. Upon LKB1 loss, it disassociates from MAT2A and reduces its enzymatic activity, leading to decreased SAM levels and subsequent induction of DNA hypomethylation. Exploring the therapeutic potential of TET inhibition to target DNA hypomethylation associated with DNPC, the authors established that pharmacological inhibition of DNA hypomethylation effectively suppresses tumor growth. These findings open new avenues for developing targeted therapies aimed at AR-independent lineage plasticity and DNA hypomethylation in DNPC.
This study advances our understanding of the molecular mechanism behind ADT-induced lineage plasticity and DNPC. These findings provide a novel mechanism by which LKB1 inactivation induces lineage plasticity and differentiation to DNPC through alteration of the metabolome and epigenome, and reveals another face of LKB1. In ARPC, LKB1 interacts with MAT2A to enhance its activity and regulate DNA methylation by harnessing the metabolome, specifically SAM as the building block for methylation. When LKB1 is lost, this interaction is disrupted, leading to alterations in the DNA methylome and induction of lineage plasticity. Hence, LKB1 inactivation drives epigenetic rewiring and tumor progression by inducing DNA hypomethylation, highlighting the complex relationship between the metabolome and epigenome in influencing lineage infidelity.
It is important to note that the shift in the metabolome can impact the epigenetic landscape beyond DNA methylation. Therefore, the impact of LKB1 inactivation on chromatin accessibility and histone modifications can be further explored. For instance, the effect of LKB1 inactivation on H3K27ac to identify super-enhancers associated with DNPC lineage determination and H3K4me3 to identify active promotors supporting lineage specification.
Integrating metabolomic and epigenomic characteristics following LKB1 inactivation exposes potential therapeutic vulnerabilities that could lead to more effective treatments to prevent or reverse the progression to treatment resistance and DNPC. These findings nominate DNA hypomethylation as an epigenetic vulnerability of DNPC, suggesting that targeting DNA hypomethylation and leveraging TET inhibitors could represent a novel therapeutic strategy for managing DNPC.
留言 (0)