
記住我
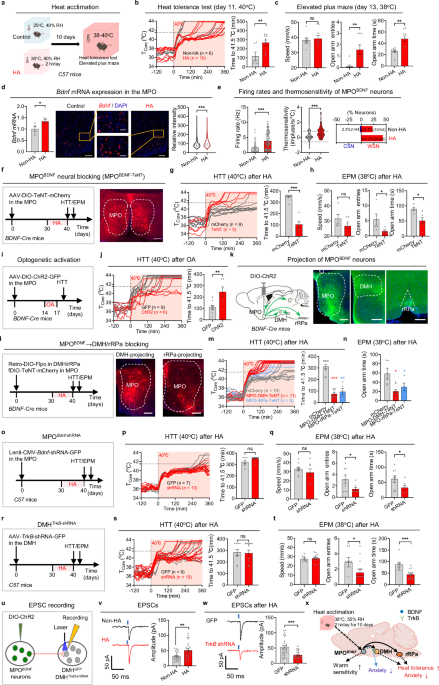
C57BL/6 (000664), Tau−/− (007251),30GRf/f (021021)28 and Rosa26a-CreERT2 (008463)29 mice were obtained from The Jackson Laboratory. GRf/f mice were crossed with Rosa26a-CreERT2 mice to generate inducible GR knockout mice. For activation of CreERT2 in adult mice, 150 mg/kg body weight of tamoxifen (Sigma-Aldrich) in sunflower seed oil (Sigma-Aldrich) was intraperitoneally injected into 10-week-old mice once a day for 5 consecutive days. Knockout efficiencies were analyzed 1 week after the last injection by qRT-PCR. Tau−/− and WT mice were littermates generated from heterozygous interbreeding. All genetic models were on the C57BL/6 background, and sex- and age-matched littermates were used for experiments, typically at 3 months of age. All animals were housed on a 12-h light-dark cycle with ad libitum access to food and water in a specific pathogen-free environment. Animal protocols were approved by Institutional Animal Care and Use Committee (IACUC) of New York University Grossman School of Medicine (#160605).
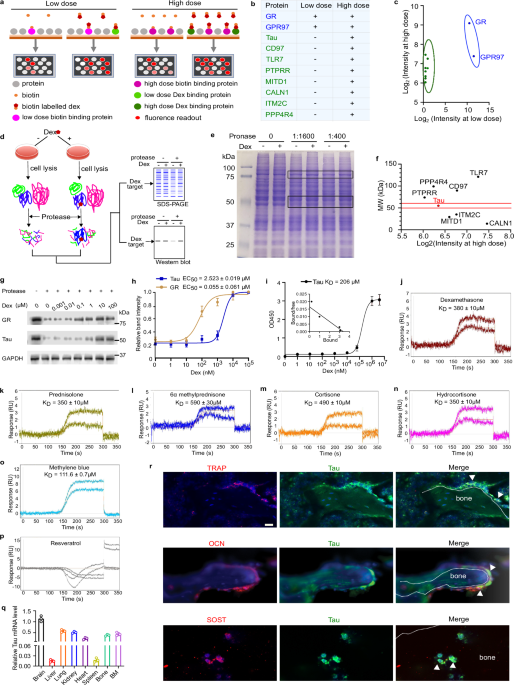
Identification of dexamethasone binding proteins using human proteome microarrayHuProt human proteome microarray version 4.0 (HuProtTM, CDI Laboratories), which is composed of ~ 20,000 human FL proteins with N-terminal glutathione S-transferase tag was used to isolate dexamethasone-binding proteins. Briefly, after blocking with 20 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.1% Tween-20, 5% BSA on an orbital shaker at room temperature for ≥ 2 h, the protein chips were incubated with 0.2 and 400 µM biotin-labeled dexamethasone, respectively, overnight at 4 °C. The next day, the protein chips were rinsed three times with phosphate-buffered saline (PBST) (with 0.1% Tween-20), followed by incubation with Cy5-conjugated streptavidin (1:1000, Invitrogen, Camarillo, CA, USA) in the dark at room temperature for 1.5 h. After washing three times with 1× PBST and then three times with Milli-Q water, the protein chips were centrifuged for 5 min in a 50 mL centrifuge tube. Finally, the protein chips were scanned with an Axon GenePix 4000B Microarray Scanner (Molecular Devices), and the probe signals were acquired using GenePix Pro6.0 software (Molecular Devices). The probes were considered detectable when the z-scores for both duplicates were greater than 2.8.
PlasmidsThe cDNA encoding FL Tau was amplified with specific primers 5’-atcgggatccgctgagccccgccaggagttc-3’containing HindIII restriction site and 5’-attatgtcgaccaaaccctgcttggccaggga-3’ containing SalI restriction site using Tau cDNA (Origene) as a template. The digested sequence was cloned into pCMV-tag2b vector (Agilent). Expression plasmids for serial FLAG-tagged deletion mutants of tau were also constructed in the pCMV-tag2b vector. The DNA sequences were confirmed by DNA sequencing performed by Psomagen.
Site-directed mutagenesisEight residues at Asp193, Ser202, Thr205, Thr212, Lys225, Ser238, Ser396, and Ser422 of Tau were individually mutated using Site-Directed Mutagenesis Kit (Agilent, #200522) in accordance with the manufacturer’s protocol. Specifically, Asp at residue 193 was mutated to Glu, Ser at residue 202 was mutated to Phe, Thr at residue 205 was mutated to Ile, Thr at residue at 212 was mutated to Asn, Lys at residue 225 was mutated to Asn, Ser at residue 238 was mutated to Tyr, Ser at residue 396 was mutated to Pro, and Ser at residue 422 was mutated to Pro, respectively.
DARTS assayDARTs assay was performed as previously described.74 For determining the molecular targets which were protected from proteinase-mediated degradation, intact Raw264.7 cells were treated with 10 µM dexamethasone, lysates were extracted with M-PER™ Mammalian Protein Extraction Reagent (Invitrogen) and subjected to protease (Sigma) digestion for 15 min at room temperature. Protease inhibitor cocktail was added to lysates followed by a 10 min incubation on ice to stop the digestion. The mixtures were separated on SDS-PAGE gel, followed by Coomassie staining.
For determining the dose-dependent curve for Tau and GR, Raw264.7 cells were lysed with M-PER™ Mammalian Protein Extraction Reagent, and incubated with serial doses of dexamethasone for 1 h with gentle rotation at room temperature. Then the mixtures were digested with pronase and subjected to western blot analysis with anti-Tau and anti-GR antibodies.
For determining the amino acid critical for dexamethasone binding, HEK293T cells were transfected with FL Tau, serial deletion Tau mutants or site-mutated Tau expression plasmids using lipofectamine 2000 according to the manufacturer’s protocol. 48 h after transfection, lysate was extracted by M-PER™ Mammalian Protein Extraction Reagent and mixed with 10 µM dexamethasone for 1 h with rotation at room temperature. Pronase (Sigma) at different concentrations was added in the reaction for 15 min at room temperature. Protease inhibitor cocktail was added to stop the digestion by incubation with the mixture on ice for 10 min. Samples were boiled in SDS loading buffer for immunoblot detection with anti-FLAG antibody.
Solid phase bindingTo compare the binding affinity between dexamethasone and Tau in vitro, microtiter plates were coated with 500 ng of Tau (SAE0076, Sigma-Aldrich) in 100 μL of Tris-buffered saline (TBS) buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.4) overnight at 4 °C. Wells were blocked using 1% bovine serum albumin (BSA) in TBS buffer for 2 h at 37 °C. After washing with TBS and 0.05% Tween, various amounts of biotin-labeled dexamethasone (sc-499756; Santa Cruz) were added. Bound protein from the liquid phase was detected using streptavidin-HRP (1:500, SA10001, Invitrogen) for 30 min at room temperature. The plates were washed with Tris-buffered saline with Tween 20 (TBST) 6 times and the reaction was visualized following a 15 min incubation with substrate 3,3’,5,5’-Tetramethylbenzidine (002023, Invitrogen). Then the signal development was stopped by H2SO4. The absorbance at 450 nm was measured by SpectraMax i3X microplate reader (Molecular Devices, LLC., CA, USA).
Molecular docking simulationsMaestro v11.1 (Schrodinger LLC, MA, USA) was used for performing molecular docking simulations. The structures of prednisolone and dexamethasone were downloaded from PubChem database. The energy of the ligand structures were minimized in Macromodel v11.5 module using OPLS3 force field, followed by a ligand preparation process in LigPrep v4.1 module according to the following parameters to generate low-energy 3D structure: different protonation states at pH 7.0 ± 2.0, and all possible tautomers and ring conformations. The prepared ligand was subjected to dock into the Tau protein model. The Tau protein structure (PDBDEV_ 00000033)75 was prepared using the Protein Preparation Wizard implemented in Maestro v11.5. The docking grid (length: 35 Å) was generated by selecting residues 150–244 as centroid. Flexible docking was performed by Glide v7.4 extra precision (XP) method. To further get an optimal simulation of binding, the best-docked pose from Glide XP docking was subjected to induced-fit docking. The docked conformations of the tested ligand were ranked based on docking score, glide energy, as well as by analyzing the binding site interactions, and the best docked pose of ligand–protein complex was selected for graphical analysis.
SPR analysisTo further determine the binding affinity between GCs and Tau, SPR analysis was performed by Essai Sciences LLC (Stillwater, OK). All experiments were carried out on the SensiQ Pioneer FE SPR platform. The buffer used was 10 mM HEPES, pH 7.4, 150 mM NaCl, and .01% Tween-20. Channels 1, 2, and 3 were activated by injection of a solution of 200 mM EDC and 50 mM NHS for six minutes. Tau, in 10 mM sodium acetate, pH 5.5, was then injected over channels 1 and 2, with channel 3 left activated, but empty, as a reference. All three channels were then injected with 1 M ethanolamine, pH 8.0, for 2 min. Small molecule analytes were dissolved in DMSO at a concentration of 10 mM. Analytes were further diluted in buffer to concentrations of 200 μM and 40 μM, at 4% DMSO. Experimental assays used the OneStep dynamic injection method which employs an analyte concentration gradient generated during injection using an analyte dispersion method. This method is particularly useful for small molecule analysis where possible molecular affinity toward immobilized target is unknown, and allows for less analyte consumption and faster assay results which are comparable to fixed concentration injection series.
Generation of Tau, GR knockout cell lines using CRISPR-Cas9To generate Tau or GR knockout Raw264.7 cells, target sgRNA oligonucleotides were annealed and cloned into BSMB1 restriction sites of a lentiCRISPRv2 vector (52961, Addgene). The sequence for each sgRNA is as follows: 5’-GCGGAGACACCCCGAACCAGG-3’ and 5’-GTGGAGCGGAGGAACCAGGGT-3’ for mouse Tau; 5’-GTGGACTTGTATAAAACCCTG-3’ and 5’-GTGGTACATCTGTCCTCCAG-3’ for mouse GR. Co-transfection of CRISPR plasmid, psPAX2 (12260, Addgene), and pMD2.G (12259, Addgene) into HEK293T (ATCC) was performed with Lipofectamine 2000 to produce the lentivirus. Then Raw264.7 cells were infected with the collected lentivirus for 18 h, followed by selection with 2 μg/mL puromycin (Gibco) for 2 days. Cells were expanded and evaluated for knockout efficiency by western blot. Two or three distinct clones for Tau or GR knockout cell lines were established, and the results from representative clone were presented.
To generate Tau or GR knockout THP-1 cells, target sgRNA oligonucleotides were annealed and cloned into BSMB1 restriction sites of a lentiCRISPRv2-mCherry vector (99154, Addgene) and a lentiCRISPRv2-GFP vector (82416, Addgene). The sequence for each sgRNA is as follows: 5’-GTGCTAAGAGCACTCCAACAG-3’ and 5’-GAAACGAAGATCGCCACACCG-3’ for human Tau; 5’-GTGGTACATCTGTCCTCCAG-3’ and 5’-GCTTTAAGTCTGTTTCCCCCG-3’ for human GR. For electroporation, the entire supplement is mixed with the Nucleofector Solution from Cell Line Nucleofector™ Kit V (VCA-1003, Lonza), and 2 µg plasmid DNA is added to 100 µL room temperature Nucleofector Solution. 2 × 106 THP-1 cells were collected by centrifugation with resuspension of the cell pellet carried out in 100 µL room temperature Nucleofector Solution and DNA mixture. The cell/DNA suspension was transferred into the cuvette and applied with the Nucleofector Program X-001 using NucleofectorTM System.76 The transfected cells were cultured for 48 h prior to sorting the successful transfectants according to mCherry signal and the cells were prepared for single-cell cloning through fluorescence-activated cell sorting. Clones were expanded and evaluated for knockout efficiency by western blot. Two or three distinct clones for Tau, GR, or Tau and GR double knockout cell lines were established, and the results from one representative one clone was presented.
IP and immunoblot analysisFor IP endogenous protein, 1 × 108 Raw264.7 cells treated with 10 µM dexamethasone for 30 min were collected and lysed with 750 µL ice-cold lysis buffer (phosphate-buffered saline, pH 7.4, 1.5% (v/w) Triton X-100, 1 mM Na3VO4, 1 mM NaF, 1 mM DTT, with protease inhibitor cocktail) on ice for 15 min, followed by sonication and centrifugation at 4 °C for 15 min. For IP TTBK1, 2 µg anti-Tau antibody (MA5-12808, Invitrogen) or mouse IgG (sc-3795, Santa Cruz) was bound to 45 µL protein A/G agarose (sc-2003, Santa Cruz) and then added to the cleared cell lysate and incubated with rotation overnight at 4 °C. For IP p105/p50, 2 µg anti-tau antibody or mouse IgG was immobilized onto magnetic beads using Pierce™ Crosslink Magnetic IP Kit (88805, ThermoFisher Scientific) and then added to the cleared cell lysate and incubated with rotation overnight at 4 °C. Immunoprecipitates were washed three times with lysis buffer and mixed with an equal volume of 2× SDS sample buffer, followed by immunoblot analysis.
For IP of ectopically expressed FL and mutated Tau, 1 × 108 Raw264.7 cells were transfected with different expression plasmids. At 48 h after transfection, cells were treated with 10 µM dexamethasone for 30 min and lysed with 750 µL ice-cold lysis buffer on ice for 15 min, followed by sonication and centrifugation at 4 °C for 15 min. Anti-FLAG M2 affinity Gel (A2220, Sigma) was added into the cleared cell lysate and incubated with rotation overnight at 4 °C. The beads were washed with lysis buffer 5 times at 4 °C and then mixed with an equal volume of 2× SDS sample buffer, followed by immunoblot analysis of TTBK1 and p105/p50.
For immunoblot analysis, the protein samples were resolved on 8% or 10% resolving gel and transferred to 0.45-µm nitrocellulose membrane (1620097, Bio-Rad) using a wet transfer system. Proteins were detected by incubation with 1:1000 dilutions of primary antibodies, washed and incubated with appropriate secondary antibodies, and detected after incubation with Western Lightning™ Plus Chemiluminescence Reagent (NEL104001, ThermoFisher Scientific) with ChemiDoc Imaging System (17001401, Bio-Rad). The following antibodies were used for immunoblotting: anti-Tau (MA5-12808, Invitrogen), anti-GR (MAI-510, ThermoFisher Scientific), anti-TTBK1 (PA5-20686, ThermoFisher Scientific), anti-p105/p50 (12540 S, Cell Signaling technology), anti-Tau pSer202/Thr205 (MN1020, ThermoFisher Scientific), anti-Tau pSer396 (44-752 G, ThermoFisher Scientific), anti-Tau Ser422 (44-764 G, ThermoFisher Scientific), anti-GAPDH (60004-I-Ig, Proteintech), anti-FLAG (F7425, Sigma), anti-CTCF (2899 S, Cell Signaling Technology), horseradish peroxidase conjugated anti-mouse IgG (115-035-003, Jackson ImmunoResearch Laboratories), and horseradish peroxidase conjugated anti-mouse IgG (111-035-003, Jackson ImmunoResearch Laboratories). Quantification of bands was performed using ImageJ software.
Biochemical co-purification and mass spectrometryTo isolate the kinases responsible for phosphorylating Tau, control and Tau knockout Raw264.7 cells were treated with or without 10 µM biotin labelled dexamethasone for 30 min, and lyzed with lysis buffer (20 mM potassium phosphate buffer, 0.15 M NaCl, pH 7.5, with proteinase inhibitor cocktail). After centrifugation, the supernatant was incubated with Streptavidin Magnetic Particles (11641778001, Sigma) for 2 h at 4 °C. The magnetic particles were then washed with lysis buffer 5 times, followed by elution with 0.1 M glycine-HCl, pH 2.5. These samples were then analyzed by mass spectrometry, performed by NYU Proteomics Laboratory. All MS/MS spectra were collected using the following instrument parameters: resolution of 15,000, AGC target of 5e4, maximum ion time of 120 ms, 1 microscan, 2 m/z isolation window, fixed first mass of 150 m/z, and NCE of 27. MS/MS spectra were searched against a Uniprot Human database using Sequest within Proteome Discoverer 1.4.
To isolate the transcription factors downstream of dexamethasone-bound Tau, Raw264.7 cells were transfected with FLAG or FLAG-tagged Tau. Forty-eight hours post transfection, cells were treated with 10 µM dexamethasone for 30 min. FLAG or FLAG-tagged Tau was affinity-purified on FLAG M2 affinity gel, and the mixture was eluted with FLAG peptide.
The eluted samples were then analyzed by mass spectrometry, performed by NYU Proteomics Laboratory. All MS/MS spectra were collected using the following instrument parameters: resolution of 15,000, AGC target of 5e4, maximum ion time of 120 ms, 1 microscan, 2 m/z isolation window, fixed first mass of 150 m/z, and NCE of 27. MS/MS spectra were searched against a Uniprot Human database using Sequest within Proteome Discoverer 1.4.
Cell culture and treatmentThe human monocytic THP-1 cells, murine macrophage RAW264.7 cells, and HEK293T cells were obtained from ATCC. RAW264.7 cells and HEK293T cells were maintained in DMEM with 10% FBS and 1% antibiotics (1% penicillin-streptomycin (15140-122, ThermoFisher Scientific)) at 37 °C under 5% CO2 in a humidified incubator. THP-1 cells were maintained in RPMI-1640 with 10% FBS and 1% penicillin-streptomycin in a humidified atmosphere of 5% CO2 in a humidified incubator.
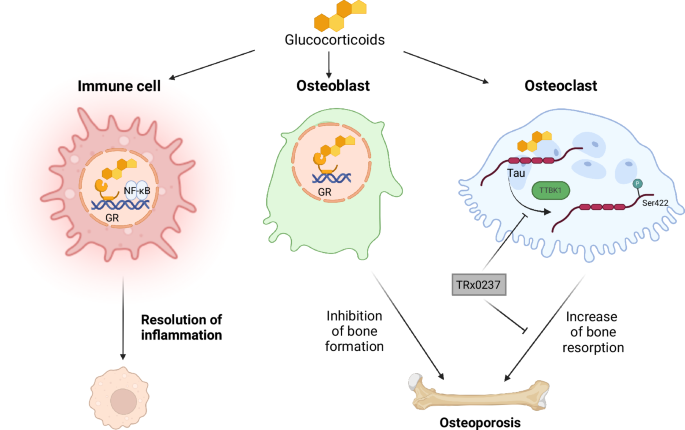
To determine the effect of dexamethasone on phosphorylation status of Tau, RAW264.7 cells were serum-starved overnight in DMEM medium, then stimulated with dexamethasone for the indicated time. To determine which kinase is responsible for phosphorylating Tau Ser422, RAW264.7 cells were pre-treated with 10 nM serine/threonine protein kinase TAO1 inhibitor (TAO kinase inhibitor) (HY-112136, MedChemExpress), 10 nM serine/threonine protein kinase Nek9 inhibitor (Dabrafenib) (HY-14660, MedChemExpress), 5 nM leucine-rich repeat serine/threonine protein kinase 2 inhibitor (MLi-2) (HY-100411, MedChemExpress), 10 mM GSK-3β inhibitor (LiCl) (7447-41-8, ThermoFisher Scientific) or 10 μM p50 inhibitor (Andrographolide) (HY-N0191, MedChemExpress) for 1 h prior to treatment with 10 µM dexamethasone for 30 min for western blot analysis or differentiation into osteoclast with 50 ng/mL RANKL and 10 µM dexamethasone for 5 days.
To knock down TTBK1 and CTCF in Raw264.7 cells, cells were transfected with TTBK1 siRNA (sc-154747, Santa Cruz), CTCF siRNA (SR419676, Origene) or scramble negative control siRNA (SR30004, Origene) using Lipofectamine 2000 (11668019, Invitrogen) for 48 h. After transfection, the knockdown efficiency was determined by western blot.
Immunofluorescence staining and confocal microscopyFor the immunofluorescence staining of total Tau, phospho-Tau Ser422, and tubulin, Raw264.7 cells plated in the 8-well chambers (Z734853, Sigma) were treated with PBS, 100 ng/mL RANKL (390-TN/CF, R&D Systems), and 100 ng/mL RANKL plus 10 µM dexamethasone for 15 min. The cells were then fixed with 100% methanol (for total Tau) or 100% acetone (for phospho-Tau Ser422), for 10 min at room temperature, followed by permeabilization in 0.1% Triton X-100 for 10 min before blocking in 1% BSA for 1 h at room temperature. The following primary antibodies were incubated with cells at 4 °C overnight: Tau antibody (1:100, MA5-12808, Invitrogen); phospho-Tau Ser422 antibody conjugated with Alexa Fluor® 647 (1:100, ITA0917, G-Biosciences). For total Tau, the cells were incubated with Alexa Fluor® 647 anti-mouse IgG (1:200, A-21235, Invitrogen) for 1 h at room temperature in the dark. Next, cells were incubated with anti-α-Tubulin antibody conjugated with Alexa Fluor® 488 (1:200, 16-232, Millipore Sigma) for 2 h at room temperature. DAPI (1:1000, D1306, ThermoFisher Scientific) was used to stain the nuclei for 10 min at room temperature.
For the immunofluorescence staining of p105/50, RAW264.7 cells grown on 8-well chambers were treated with PBS, 100 ng/mL RANKL, or 100 ng/mLRANKL plus 10 µM dexamethasone for 4 h. After treatment, the cells were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature, then permeabilized in 0.1% Triton X-100 for 10 min, followed by blocked in 1% BSA for 1 h at room temperature. Cells were then incubated with anti-NF-κB p105/p50 antibody (1:100, 13586 S, Cell Signaling Technology) at 4 °C overnight, followed by anti-Rabbit IgG conjugated with Alexa Fluor® 488 (1:200, A-11008, Invitrogen) for 1 h at room temperature. DAPI (1:1000) was used to stain the nuclei for 10 min at room temperature.
For the immunofluorescence staining of total Tau and TTBK1, Raw264.7 cells plated in the 8-well chambers were treated with or without 10 µM dexamethasone for 30 min. Cells were fixed with 4% paraformaldehyde in PBS pH 7.4 for 10 min at room temperature, then permeabilized using 0.1% TritonX-100 for 10 min at room temperature. Cells were blocked by incubating with 5% BSA for 1 h at room temperature. Immunofluorescence staining was performed by using anti-Tau antibody (1:100, MA5-12808, ThermoFisher Scientific) and anti-TTBK1 antibody (1:100ABN348, Millipore). These primary antibodies were detected with Alexa Fluor 488-labeled goat anti-mouse IgG (1:200, A-11001, ThermoFisher Scientific) and Alexa Fluor 546-labelled goat anti-rabbit IgG (1:200, A-10040 ThermoFisher Scientific). DAPI (1:1000) was used to stain the nuclei for 10 min at room temperature. All images were taken on a Zeiss laser scanning microscope (LSM) 780 with a 63×, 1.4 NA oil objective. Fluorescence intensity was quantified using ImageJ software.
Osteoblastogenesis and osteoclastogenesisThe bone marrow cells isolated from 12-week-old male WT, Tau−/−, and GR−/− mice were cultured in MEM-α with 10% FBS and 1% penicillin-streptomycin (15140-122, ThermoFisher Scientific) for at least 24 h. For osteoblastogenesis, the adherent cells were differentiated in osteogenic induction medium (10% FBS, 100 µg/mL ascorbic acid and 10 mM β-glycerophosphate in MEM-α) in the absence or presence of 10 nM or 10 µM dexamethasone. For osteoclastogenesis, the non-adherent cells were cultured with 20 ng/mL M-CSF (576406, Biolegend) for 3 days, and switched to the differentiation medium including 20 ng/mL M-CSF, 50 ng/mL RANKL (390-TN-010/CF, R&D Systems), with or without 10 nM or 10 µM dexamethasone. To differentiate Raw264.7 cells to osteoclasts, cells were treated with 50 ng/ml RANKL in the presence or absence of 10 µM dexamethasone. To differentiate THP-1 cells to osteoclasts,77 the cells were incubated with 100 ng/mL phorbol‑12 myristate‑13 acetate for 48 h to differentiate into adherent macrophages, and subsequently, 50 ng/mL RANKL and 20 ng/mL M-CSF in the presence and absence of 10 µM dexamethasone were added into the adherent cells. The medium was replaced every day.
After differentiation into osteoblasts for 10 days and osteoclasts for 5 days, respectively, cell apoptosis was detected using In Situ Cell Death Detection Kit (11684795910, Sigma) in accordance with the manufacturer’s instructions. Briefly, the cells were fixed with a freshly prepared fixation solution (4% paraformaldehyde in PBS, pH 7.4) for 1 h at room temperature. After rinsing with PBS, the cells were then incubated with permeabilization solution (0.1% Triton X-100 in 0.1% sodium citrate) for 2 min on ice. After washing twice with PBS, 50 μL of TUNEL reaction mixture was added. Then the cells were incubated in a humidified atmosphere for 1 h at 37 °C in the dark. DAPI (1:1000) was used to stain the nuclei for 10 min at room temperature. The samples were then analyzed under the confocal microscope.
After differentiation into osteoblasts for 21 days from primary bone marrow cells, cells were fixed in 4% PFA and stained with Alizarin Red S (A5533-25G, Sigma-Aldrich). At the end of differentiation into osteoclasts from bone marrow cells for 7 days, Raw264.7 cells for 5 days or THP-1 cells for 14 days, cells were stained for osteoclasts using the TRAP kit (387 A, Sigma-Aldrich), the number of TRAP-positive multinucleated cells containing more than 3 nuclei and the number of nuclei per osteoclast were counted using microscopy.
Bone resorption activity was measured using a fluoresceinated calcium phosphate-coated OsteoAssay plates. The coated calcium phosphate is first bound to fluoresceinamine-labeled chondroitin sulfate, which is released from the calcium phosphate layer into conditioned medium by osteoclastic resorption activity. To reveal osteoclast resorption pits, bone marrow cells grown on OsteoAssay plate (CSR-BRA-48, Cosmo Bio Co., Ltd) were stimulated with 20 ng/mL M-CSF, 100 ng/mL RANKL with or without 10 µM dexamethasone for 7 days. At the end of stimulation, the conditioned medium with fluoresceinamine-labeled chondroitin sulfate from each well was transferred to 96-well black plate and mixed with bone resorption assay buffer in accordance with the manufacture’s guidelines. The fluorescence intensity of each well was detected with an excitation wavelength of 485 nm and an emission wavelength of 535 nm. Cells were then removed by sodium hypochlorite (5%) for 5 min. The bottom of the well was photographed by a light microscope, and the resorption area per well was calculated by ImageJ software.
Primary osteocyte isolationOsteocytes were isolated from mouse long bones using a method described by ref.78 Briefly, tibiae and femurs were aseptically dissected, and bone marrow was removed. The bones from 3 different mice with the same genetic background were pooled together and subjected to nine sequential digestions to remove periosteum, fibroblast, osteoblast, osteoclast and other adherent cells. Collagenase solution (0.25 mg/mL collagenase type I and 0.75 mg/mL collagenase type II) was prepared in αMEM, and EDTA solution (5 mM) was prepared in magnesium and calcium-free PBS with 1% BSA.
Quantitative RT-PCRTotal RNA was isolated from cultured cells or pulverized tibia using RNeasy Mini Kit (74106, Qiagen). cDNA was prepared using 1 µg RNA with the High-Capacity cDNA Reverse Transcription Kit (4368813, Applied Biosystems). SYBR green-based (4367659, Applied Biosystems) quantitative PCR was performed in triplicate on the Real-Time PCR System (Applied Biosystems) using the following primers: NFATc1 forward 5’- CATCCTGTCCAACACCAAAGTC-3’ and reverse 5’-GTGTTCTTCCTCCCGATGTCTG; CTSK forward 5’-ATGGAAGAAGACTCACCAGAAG-3’ and reverse 5’-CCACTTCTTCACTGGTCATGTC-3’; CTR forward 5’- GTCTTGCAACTACTTCTGGATGC-3’ and reverse 5’-CGTGGATAATGGTTGGCACTATC-3’; TRAP forward 5’- CAGCAGCCAAGGAGGACTAC-3’ and reverse 5’-CACATAGCCCACACCGTTCTC-3’; OPG forward 5’-TTGCCTGGGACCAAAGTGAATG-3’ and reverse 5’-GCTGCTTTCACAGAGGTCAATG; Runx2 forward 5’-ATGCTTCATTCGCCTCACAAA and reverse 5’-ATGCTTCATTCGCCTCACAAA; OCN forward 5’-CTGACCTCACAGATCCCAAGC and reverse 5’-TGGTCTGATAGCTCGTCACAAG; Col1a forward 5’-TGAACGTGGTGTACAAGGTC-3’ and reverse 5’-CCATCTTTACCAGGAGAACCAT; BSP forward 5’-AAGCACAGACTTTTGAGTTAGC-3’ and reverse 5’-ACTTCTGCTTCTTCGTTCTCAT; Gapdh forward 5’-CCCAGAACATCATCCCTGCATC-3’ and reverse 5’-TCTTGATGTCATCATACTTGGCAG-3’. mRNA levels were normalized to Gapdh and reported as relative mRNA fold change.
ELISA analysisELISA-based assay was performed to detect the inhibitory effects of FDA-approved drugs on dexamethasone-dependent p-Tau Ser422. Briefly, ELISA plates were coated with 12.5 ng anti-Tau antibody (MA5-12808, Invitrogen) diluted in PBS overnight at 4 °C and blocked with 5% BSA in PBS for 2 h at room temperature. THP-1 cells were serum-starved overnight in RMPI-1640 medium, then pre-treated with or without individual drug in an FDA approved drug library (L1300, Selleckchem) for 1 h prior to stimulation with 10 µM dexamethasone for 30 min. Cells were lyzed with lysis buffer (50 mM HEPES, 10 mM MgCl2, 1% SDS, 150 mM NaCl with protease and phosphatase inhibitor cocktail), the subsequent supernatant was collected after centrifugation and protein concentration was determined using Pierce™ BCA Protein Assay Kit (23225, ThermoFisher Scientific). The standard curve was prepared by adding serial dilution of the supernatant collected from dexamethasone-treated THP-1 cells with a protein concentration of 0–400 ng/mL into the plates and the incubation with anti-Tau antibody coated-plates at room temperature for 2 h. Alternatively, for inhibition of dexamethasone-dependent activation of Tau phosphorylation at Ser422 by drugs, 200 ng/mL supernatant collected from individual library drug plus dexamethasone-treated cells was added into the plates and incubated for 2 h at room temperature. After washing, 12.5 µg/mL biotinylated phosphor-Tau antibody was then added into the plates and incubated for another 2 h at room temperature. After washing with PBST 6 times, the bound protein-antibody mixture was detected using streptavidin-HRP (1:500, SA10001, Invitrogen) for 30 min at room temperature. The plates were washed with PBST 6 times and the reaction was visualized by following 15 min incubation with substrate 3,3’,5,5’-Tetramethylbenzidine (002023, Invitrogen) with signal development stopped by H2SO4. The absorbance at 450 nm was measured by a microplate reader.
ELISA kits were used to detect levels of RANKL (MTR00, R&D Systems), OPG (MOP00, R&D Systems), CTX-1 (MBS9141384, MyBiosource), PINP (MBS2500076, MyBiosource) in sera collected from indicated murine models in accordance with the manufacturer’s instruction.
ChIP-qPCRRaw264.7 cells were treated with or without 50 ng/mL RANKL for 1 h, followed by fixation with 1% formaldehyde for 10 min at room temperature. ChIP was performed using antibodies against p50 and p65, following the manufacture instructions (ThermoFisher Scientific). The purified DNA was analyzed by PCR with primers specific to the NFATc1 promoter: GGACGCCCATGCAATCTGTTAG (sense) and GTGCCCTGAGAAAGCTACTCTC (antisense).79 qPCR results from immunoprecipitated samples were normalized to that from input DNA.
GIO modelTwelve-week-old male and female WT, Tau−/− and GR−/− mice received intraperitoneal injections of PBS or 10 mg/kg body weight dexamethasone (D2915, Millipore Sigma) daily for five consecutive weeks. Mice were then sacrificed, and long bones (tibia and femur), vertebrae (L1 and L2), and sera were collected for further study.
To determine the therapeutic effect of TRx0237 on dexamethasone-induced bone loss, twelve-week-old male WT, Tau−/−, and GR−/− mice were intraperitoneally injected with PBS, 10 mg/kg body weight dexamethasone, or 10 mg/kg body weight dexamethasone plus 4 mg/kg body weight TRx0237 (S7762, Selleckchem) daily for five consecutive weeks. After treatment, all mice were sacrificed, and long bones (tibia and femur), vertebrae (L1 and L2), and sera were collected for further study.
CIA induction and assessmentTwelve-week-old WT, Tau−/−, and GR−/− male mice were immunized intradermally with 100 μL emulsion containing an equal volume of chicken type II collagen (20012, Chondrex) and complete Freund’s adjuvant (7001, Chondrex) at the base of the tail (day 0). Booster injections were performed with chicken type II collagen emulsified in incomplete Freund’s adjuvant (7002, Chondrex) on day 19. Seven days after booster injections (day 26), the mice with CIA phenotype were intraperitoneally injected with PBS or 10 mg/kg body weight dexamethasone daily for 5 consecutive weeks. To determine the therapeutic effect of TRx0237 on dexamethasone-induced bone loss in CIA mice, WT mice with CIA phenotype were given daily intraperitoneal injections of PBS, dexamethasone (10 mg/kg body weight), or dexamethasone (10 mg/kg body weight) plus TRx0237 (4 mg/kg body weight) for 5 consecutive weeks. Meanwhile, the clinical score was assessed individually every other day until euthanasia using the following system: 0 = no erythema and swelling; 1 = erythema and mild swelling confined to the tarsals or ankle joint; 2 = erythema and mild swelling extending from the ankle to the tarsals; 3 = erythema and moderate swelling extending from the ankle to metatarsal joints; 4 = erythema and severe swelling encompass the ankle, foot, digits, or ankylosis of the limb. Each paw was scored and scores of four paws were summed to give a total clinical score with a maximum possible score of 16 for each mouse. After five weeks of treatment, the mice were sacrificed (day 62), and long bones (tibia and femur), hind paws, and sera were harvested for further study.
DEXA scan analysisAt the end of the establishment of the mouse model, mice were anesthetized using 200 µL anesthetic agent containing 20 mg/mL ketamine and 2 mg/mL xylazine, and the whole body was scanned on a Lunar Piximus bone densitometer (GE Medical System Lunar Corp.). The instrument was calibrated before each scanning session using a Phantom with known BMD according to the manufacturer’s instruction. The bone mineral density was analyzed using Lunar Piximus 2.10 software.
µCT analysisLong bones (tibia and femur) and vertebrae (L1 and L2) from WT, Tau−/−, and GR−/− mice were fixed in 4% PFA for 24 h and then stored in 70% ethanol. The fixed bone samples were scanned using the high-resolution SkyScan microCT system (SkyScan 1172. Kontich, Belgium). The scanning method and settings are: the samples were scanned using a 10-MP digital detector, 10 W of energy (60 kV and 167 mA), and a pixel size was 9.7 microns, exposure 925 ms/frame rotation step 0.3 degrees with ×10 frame averaging, 0.5 mm Aluminum filter and scan rotation was 180 degrees. After scanning, the radiographs were reconstructed using NRecon software (version 1.7.3.0; Bruker microCT, Kontich, Belgium). Reconstruction was made with NRecon using GPU acceleration. Gaussian smoothing was applied with a 2 voxel radius, ring artefact and beam hardening corrections were applied in reconstruction. Ring artefact reduction was set to 7 pixels. Beam hardening correction was set to 40%. After reconstruction, the parameters of trabecular bone of tibia, femur, and vertebrae (L1 and L2) including BV/TV (%), Tb.Th (μm), and Tb.N (1/mm), were analyzed using CT Analyser (CTan) v.1.18.8.0 (Bruker). The 3D µCT images were obtained by CTvox v.3.3.1 software (Bruker). The region of interest was defined as follows: starting at a distance of 100 slices from the distal growth plate of the femur and extending a further distance of 200 slices in the proximal direction for the femur; starting at a distance of 50 slices from the proximal growth plate of the tibia and extending a further distance of 200 slices in the distal direction for tibia. The trabecular bone of vertebrae L1 and L2 was analyzed.
Dynamic histomorphometric analysisFor calcein labeling of non-decalcified bone specimens, mice were intraperitoneally injected with 100 µL 2 mg/mL calcein (C0875, Sigma-Aldrich) in a 2% sodium bicarbonate solution per 20 g body weight at day 8 and day 3 before euthanasia. The femoral trabecular bone was analyzed in a 1.5 cm long region of interest starting 200 µm under the mineralized front of the growth plate.
Histological analysisDissected, soft-tissue specimens were fixed in 4% PFA at 4 °C overnight, and then decalcified with 10% EDTA (pH 7.2) for 2 weeks for long bones and vertebrate, and 4 weeks for hind paws at 4 °C with a change of EDTA every other day. After samples were embedded in paraffin, 6-μm serial paraffin sections were prepared. Representative sections were stained with freshly prepared H&E and TRAP. H&E staining was performed according to the following order: two changes of xylene 2 min each; two changes of 100% ethanol 2 min each; one change of 95% ethanol for 2 min; one change of 80% ethanol for 2 min; one change of distilled water for 2 min; one change of hematoxylin solution (3801570, Leica Biosystems) for 2 min; one change of distilled water for 2 min; one change of differentiating solution (3803590, Leica Biosystems) for 2 min; one change of distilled water for 2 min; one change of blue buffer (3802915, Leica Biosystems) for 2 min; one change of distilled water for 2 min; one change of 95% ethanol for 2 min; one change of eosin solution (3801606, Leica Biosystems) for 2 min; one change of 95% ethanol for 2 min; two changes of 100% ethanol 2 min each; two changes of xylene 2 min each. The stained sections were mounted with Cytoseal XYL (83124, ThermoFisher Scientific). Inflammation scores were obtained based on the following criteria: 0 = no inflammation; 1 = slight thickening of the lining layer or some infiltrating cells in the underlying layer; 2 = slight thickening of the lining layer plus some infiltrating cells in the underlying layer; 3 = thickening of the lining layer, an influx of cells in the underlying layer, and presence of cells in the synovial space; 4 = synovium highly infiltrated with many inflammatory cells. Sections were stained for TRAP using the following protocol: two changes of xylene 2 min each; one change of 100% ethanol for 2 min; one change of 95% ethanol for 2 min; two changes of distilled water 2 min each. Then the sections were incubated with TRAP staining solution at 37 °C for 60 min and counterstained with 0.02% fast green for 2 min, followed by dehydrating through 95% ethanol, 100% ethanol, and xylene 5 s each. All images were captured using the Zeiss microscope (Axio Scope A.1, Carl Zeiss, LLC). The quantification of osteoclast surface and osteoblast surface on TRAP- and H&E-stained sections were performed with ImageJ software.
Immunohistochemistry and immunofluorescence stainingFor immunohistochemistry staining, deparaffinized and hydrated sections were incubated with 0.1% trypsin for antigen retrieval (30 min, 37 °C), followed by incubation with 3% H2O2 (30 min, 4 °C). After blocking with the solution containing 3% BSA and 20% goat serum for 60 min at 37 °C, sections were incubated with primary antibodies against phospho-Tau (Ser 422) (1:100, 44-764 G, Invitrogen) overnight at 4 °C. Then biotinylated anti-rabbit secondary antibodies (1:200, BA-1000-1.5, Vector Laboratories) were added into the sections for 60 min at room temperature. After washing with PBS, the Vectastain Elite ABC kit (PK-6100, Vector Laboratories) was used to amplify signals. Sections were then incubated with 0.5 mg/mL 3,3-diaminobenzidine in 50 mM Tris-Cl substrate (Sigma-Aldrich) until desired stain intensity developed, and counterstained with 1% methyl green.
For immunofluorescence staining, deparaffinized and hydrated sections were incubated with 0.1% trypsin for antigen retrieval (30 min, 37 °C). Subsequently, sections were blocked with 10% goat serum for 30 min, and then incubated with primary antibodies against TRAP (1:100, PA5-116970, Invitrogen), osteocalcin (1:100, M173, Takara), sclerostin (1:100, PA5-46977, Invitrogen) or Tau (1:100, 10274-1-AP, ProteinTech) overnight at 4 °C. Following this, sections were inbubated with the corresponding secondary antibodies for 1 h in the dark, followed by counterstaining with DAPI. Images were obtained using the Zeiss microscope, and the quantification was performed with ImageJ software.
Collection of human bone samplesHuman samples were obtained from patients underwent operations at Daping Hospital (Chongqing, China). In order to eliminate the effects of estrogen, all samples were collected from men patients. Osteoporosis was defined based on BMD measured by dual-energy X-ray absorptiometry (DXA, GE Health), according to standards (BMD T value <−2.5). Subjects with secondary osteoporosis, malignancy, hypertension, diabetes and other metabolic syndromes, cognition impairment were excluded from our study. Normal control human bone tissues were obtained from patients with fracture or suffered traumatic amputation (n = 5, aged from 35 to 40 years old), and bone tissues of osteoporosis patients were obtained from patients who underwent joint replacement (n = 6, aged from 60 to 80 years old). The study was performed in accordance with the Declaration of Helsinki and approved by the Ethics Committees of Daping Hospital (ID: #2019-127-01). Participants provided written informed consent to participate in the study.
Statistical analysesStatistical significance was determined using GraphPad Prism 9 (GraphPad). Statistical significance between two groups was determined using a two-tailed, unpaired Student’s t-test. For comparison of more than two groups, ANOVA analysis was used with Bonferroni post-hoc test as described in the figure legends. P values were considered significant if P < 0.05. All data points represent biological replicates, unless otherwise indicated in the figure legends. All data are presented as means ± SD unless otherwise stated in the figure legends. All specific statistical details can be found in the figure captions.
留言 (0)