
記住我
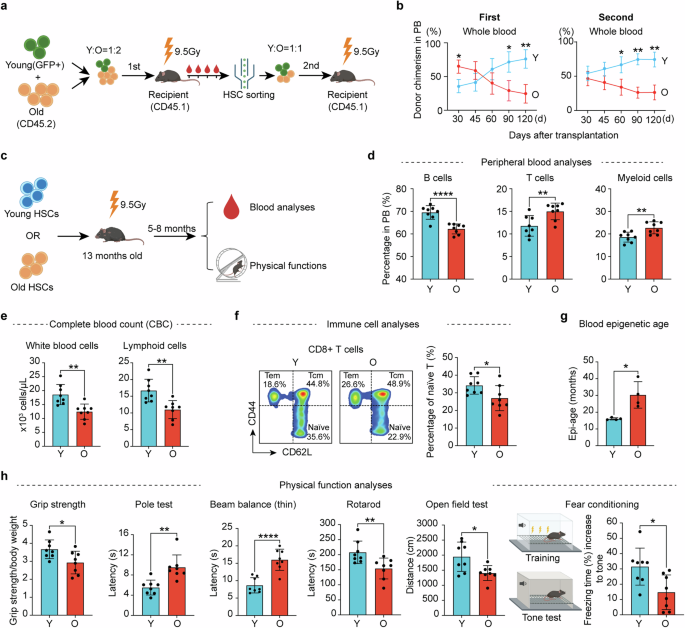
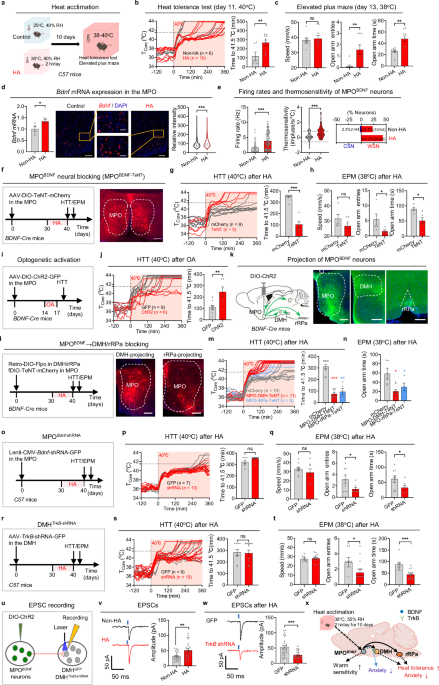
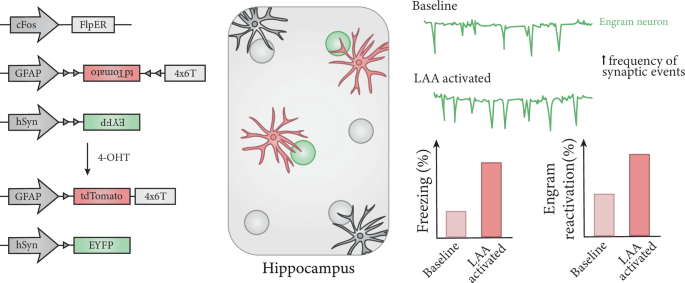
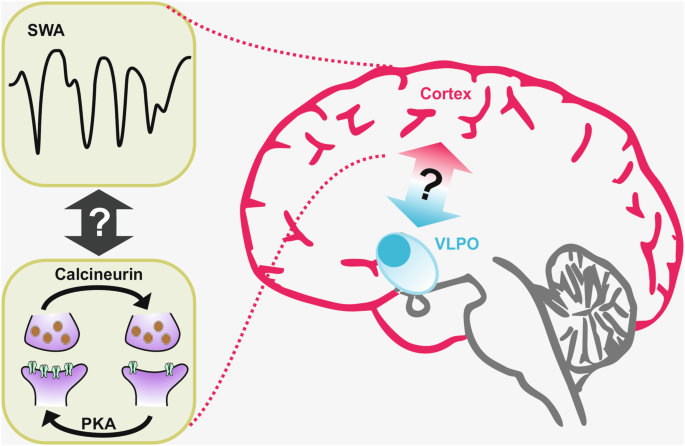
Foundation models in transcriptomics have gained attention due to their ability to generalize across tasks with limited labeled data. GeneCompass builds upon these models by incorporating prior biological knowledge and datasets from both human and mouse cells, enhancing its capacity for cross-species analysis and advancing the field of single-cell transcriptomics.
Foundation models like those in ChatGPT1 have revolutionized various fields. Based on transformer architectures and pre-trained on vast unlabeled datasets, they require extensive data to comprehend context and structure effectively. These models can tackle specific problems with little to no labeled data through zero-shot learning or fine-tuning. This is particularly relevant in biomedical research, where high-quality labeled datasets are scarce. Large language models have been quickly adopted due to their easily demonstrable performance on intuitive tasks, such as language understanding and standardized tests. However, translating this success to molecular biology involves adapting models to molecular data and establishing benchmarks demonstrating that these models understand cell biology and can predict cellular behavior.
To address this challenge, several approaches adapt these models to cells, leveraging the increasing availability of large-scale single-cell transcriptome data.2,3 In this context, genes and their expression levels are analogous to words, and each cell represents a sentence. Implementing this analogy in a concrete model raises questions: How can we efficiently encode transcriptome information? Which model architectures are optimal? How do we define and measure “best” performance? What roles do training data size and other factors play in model effectiveness?
In a recent study in Cell Research, Yang et al.4 advanced this field by proposing GeneCompass, a transformer-based foundation model trained on more than 100 million human and mouse cells — the largest single-cell corpus to date. Unlike previous models that only considered gene ranks or bins, GeneCompass quantitatively encodes absolute gene expression values and integrates prior biological knowledge, including gene regulatory networks (GRNs), promoter information, gene families, and co-expression data (Fig. 1). Through ablation studies on various downstream tasks, they disentangle the effects of input data (like prior information and corpus size) and model architecture. By evaluating several downstream tasks assessed by other models, they push toward a unified evaluation benchmark for objective assessment.
Fig. 1: Overview of transcriptomic foundation models.
GeneCompass is pre-trained on vast unlabeled datasets to effectively comprehend context and structure and then applied to specific problems with little to no labeled data through zero-shot learning or fine-tuning.
The authors applied the embeddings learned during pre-training to tasks such as cell type annotation, perturbation prediction, dosage-sensitive gene prediction, and GRN inference. By comparing various models, including GeneCompass variants, to known labels, they demonstrate the benefits of incorporating prior knowledge and large-scale pre-training. For example, models with prior knowledge outperform those without. To isolate model architecture’s impact from pre-training data, they retrained competing models on the same dataset, revealing that GeneCompass consistently outperforms them across tasks.
Scaling laws in foundation models suggest that pre-training on larger datasets can significantly boost performance. The authors demonstrate these laws across various downstream tasks, although room for improvement remains. For instance, in cell type annotation, accuracy plateaus indicate limits of the pre-training corpus. In tasks such as drug response, gene expression profiling, and dosage-sensitive gene prediction, performance increases substantially with larger pre-training data. In the latter, GeneCompass consistently outperforms Geneformer3 but plateaus at an AUC of 0.95, highlighting its architectural advantage.
In cell type annotation, GeneCompass outperforms other foundation models trained on the same data for species-specific datasets but does not consistently surpass a dedicated model in cross-species annotation, indicating a need for further development.
For GRN inference, GeneCompass leverages the relationships captured in its gene embeddings. By measuring the similarity between gene embeddings and applying a threshold to determine significant interactions, they constructed a gene–gene relationship network. This approach outperforms other foundation models and a state-of-the-art method. To validate their findings more objectively, the authors performed in-silico gene deletions and examined the predicted changes in other genes, focusing on known direct targets.
To assess drug response prediction, the embeddings were integrated with the Compositional Perturbation Autoencoder (CPA)5 framework to predict gene expression changes. GeneCompass achieved scores comparable to Geneformer, indicating that prior knowledge and a larger cross-species training corpus do not enhance performance in this task. Similar observations occur when using DeepCE6 for predicting drug-induced gene expression changes. Finally, fine-tuning GeneCompass for predicting dosage-sensitive genes yielded superior performance compared to Geneformer and models without pre-training.
For predicting the effect of gene perturbations, the embeddings are integrated into GEARS,7 an advanced perturbation prediction tool. GeneCompass embeddings improved predictions compared to default GEARS embeddings. However, this task remains challenging for deep learning models; a recent study shows that even simple linear models can outperform foundation models.8
Finally, cell fate prediction is evaluated by in-silico gene modifications to drive cells toward a target state. The authors apply this to human embryonic stem cells (ESCs) differentiating into gonadal lineage cells, identifying five transcription factors (TFs), three of which are known to play roles in mouse gonadal development in vivo. Indeed, human ESCs overexpressing NR5A1 and GATA4 exhibit characteristics of differentiated gonadal cells. This validates the practical applicability of their approach.
While foundation models are often evaluated using similar downstream tasks, aiding in direct comparisons, many of these metrics primarily serve as proof of concept. They often lack validation in practical applications, revealing a gap between theoretical performance and real-world utility. For instance, the GRN task shows promise, but to maximize its benefit, more extensive validation is needed, such as testing and adjusting to additional TFs. Moreover, a general issue with GRNs is the absence of a definitive ground truth. Other tasks like perturbation prediction remain challenging, and foundation models have only partially improved them so far. Future community efforts should focus on establishing meaningful and comparable benchmarks to convincingly demonstrate the performance and utility of foundation models.
In conclusion, GeneCompass represents a significant advancement in foundation models for single-cell transcriptomics. Importantly, it demonstrates that the model follows scaling laws in some downstream tasks, suggesting that adding more data to the training corpus could improve performance. However, merely increasing the pre-training corpus is insufficient; the new data should offer greater diversity across tissues, developmental stages, diseases, and more to be beneficial. Ultimately, developing a universal foundation model — or “virtual cell”9 — that integrates transcriptomic, epigenetic, genomic, proteomic, metabolomic, and imaging data could revolutionize our understanding of biological processes. Such a comprehensive model would enable the exploration of unknown biology and accelerate drug and biomarker discovery. While it is still unclear how and to what extent this is possible, GeneCompass is a step forward.
留言 (0)