
記住我

The TCGA database was utilized to identify differentially expressed genes between gliomas and normal tissues. The heatmap illustrates the overall gene expression in gliomas, revealing a total of 1328 differentially expressed genes (Fig. 1A). The PPI network volcano plot highlights the significance values (P-values) and fold changes of the differentially expressed genes and their interactions(Fig. 1B).
Fig. 1
Identification of DEGs in gliomas. A Heatmap of differentially expressed immune-related genes; B protein-protein interaction (PPI) combined volcano plot of genes
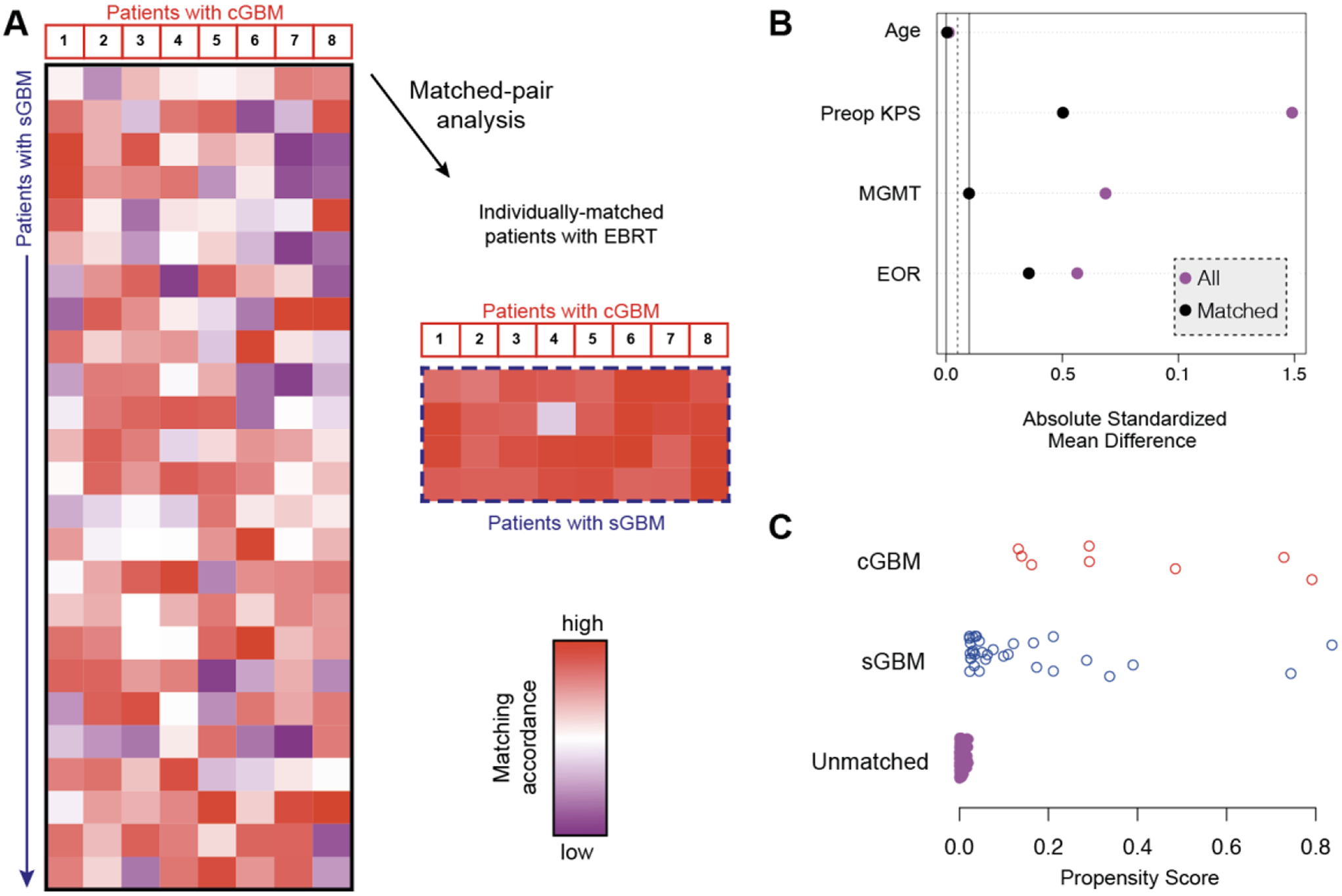
Building prognostic models based on machine learningAnalysis of the expression patterns of 1328 immune-related differentially expressed genes identified 199 potential prognostic markers via univariate Cox analysis, as depicted in Fig. 2A. Building on these findings, a comprehensive machine learning framework was crafted, leveraging to formulate a prognostic model for gliomas with an immune-related consensus. During the machine learning phase, any algorithms that predicted five or fewer genes across the 101 algorithmic combinations and had a composite index below 0.5 were eliminated, as shown in Fig. 2B. Although the RSF + plsRcox and Lasso + plsRcox algorithms yielded identical composite scores, the Lasso + plsRcox algorithm was selected as optimal due to its higher C-index in the training dataset.
Fig. 2
Developed the best prognostic model on machine learning. A Univariate Cox analysis was conducted, and B predicted 101 prognostic models
Validation of the prognostic modelTo further evaluate the clinical applicability of the model, patient samples were assigned scores according to the model and divided into high- and low-risk groups. Both univariate and multivariate analyses confirmed that the model’s risk score was a significant independent predictor of glioma prognosis, as illustrated in Fig. 3A and B, with a statistically robust association (P < 0.001). The survival analysis indicated that the low-risk group was better than others, as shown in Fig. 3C and D. The model’s risk score outperformed other prognostic indicators in predicting survival times for glioma patients, as demonstrated in Fig. 3E and F. Finally, a bar chart was created to visually represent the distribution of risk scores among the patient cohort, as seen in Fig. 3G.
Fig. 3
Clinical relevance validation of the prognostic model. A Univariate Cox analysis; B Multivariate Cox analysis; C Survival curves in the training set; D Survival curves in the test set; E Time-dependent ROC curve; F Concordance index (C-index)
Immune relevance verification and functional enrichment analysisTo investigate the model’s involvement in immune processes, an analysis utilizing various platforms was undertaken of the model and immune cells. The results indicate that differences exist in immune cells and their functions, immune checkpoints, and comprehensive scores between the two group (Fig. 4A–D). Specifically, the scores for stem cells, immune cells, and specific tumor immune assessment are higher than those in low (Fig. 4D). Firstly, the GSEA analysis was independently conducted on the high-risk and low-risk groups, unveiling notable disparities in gene expression between these two risk levels. In the high-risk group, the gene expression profile is linked to the activation of immune response and inflammation processes, whereas the low-risk group exhibits an enrichment of gene sets about neural system function and cell signal transduction (see Fig. 4E, F).
Fig. 4
Immune-related validation, i.e., functional enrichment analysis. A Immune checkpoint - box plot; B Immune cell function - box plot; C Immune cell correlation analysis across multiple platforms; D Immune infiltration - violin plot; EGSEA analysis - low-risk group; FGSEA analysis-high-risk group
Genomics of model genesTo further explore the genomics of model genes, their genetic variations and interactions were studied. Genetic mutation analysis showed that missense mutations caused by C > T SNPs were predominant in model genes. Among them, ELN, IKBKE, SSTR2, BMP2, and CXCL13 were the most prominent (Fig. 5A). In terms of copy number variations, CDK4, BIRC5, and SSTR2 were mainly associated with copy number increase, while APOBEC3C was predominantly associated with copy number loss (Fig. 5B). By categorizing the model genes into risk factors and protective factors, a correlation expression circle plot was drawn, revealing complex relationships in gene expression (Fig. 5C). Additionally, a chromosome localization circle plot of model genes was created (Fig. 5D).
Fig. 5
Genomics of Model Gene. A Tumor mutation burden of model gene; B Copy number variation of model gene; C Expression correlation network of model gene; D Chromosomal localization of model gene
Impact of a model gene at the single-cell levelTo delve deeper into the impact of model genes on gliomas, an examination will be conducted utilizing the TISCH database, we analyzed online single-cell datasets. The results indicate that model genes, validated across multiple datasets, show significant expression changes in both “Mono/Macro” (monocytes/macrophages) and “Oligodendrocyte” (oligodendrocyte) cell lineages (Fig. 6).
Fig. 6
TISCH database perspective: analysis of the role of single-cell level model genes in gliomas
Cellular localization of model genes and pathological associationsTo further explore the biological information of model genes and their expression in gliomas, model genes were searched in the HPA database. The results of immunohistochemistry with differences were displayed, along with the immunofluorescence of the model genes to observe their localization (Fig. 7). The results indicated that the differential expression of CDK4, ELN, IKBKE, NMB, SSTR2, and TGFBR1 in gliomas is associated with the pathology of the tumor. Firstly, CDK4 was found in both the cytoplasm and the nucleus. As a key kinase in cell cycle regulation, it participates in controlling the G1 to S phase transition of the cell cycle. Secondly, ELN primarily localizes in the extracellular matrix. As an important structural protein, it plays a crucial role in maintaining tissue elasticity and integrity. Additionally, IKBKE acts in both the cytoplasm and the nucleus. As a component of the NF-κB signal pathway, it influences cell immunity and stress response. Next, NMB functions as a secretory protein mainly in the neuroendocrine system, participating in intercellular signal transduction. Meanwhile, SSTR2 is positioned on the cell membrane. As a somatostatin receptor, it regulates cell growth and secretion activities. Lastly, TGFBR1 is also located on the cell membrane. As a receptor of the TGF-β signal pathway, it participates in regulating cell proliferation, differentiation, and apoptosis processes.
Fig. 7
Understanding the pathology of gliomas from a molecular perspective: immunohistochemistry and immunofluorescence research results in the HPA database
Drug network analysis and molecular docking verification of model genesTo further explore the potential of model genes in clinical applications, we conducted a drug network analysis on these genes. The data analyzed was sourced from the DGibd database, covering 16 key genes and 236 related drugs, producing a total of 250 analysis results (Fig. 8A). Further core hub analysis of the drug network was carried out to provide a basis for subsequent molecular docking verification (Fig. 8B). Considering that the DGibd database contains some predictive information, molecular docking experiments were conducted to preliminarily validate these predictions. The molecular docking results showed good docking between the selected drugs and model genes (Fig. 8C).
Fig. 8
Drug network and molecular docking. A Gene-Drug network; B central hub analysis; C molecular docking
Single-cell analysis of central hub genesIn the single-cell expression analysis, the t-SNE plot (Fig. 9A) illustrates the clustering of diverse cell types, Microglia, Proliferating glioblastoma cells, Neuronal cells, Inhibitory neurons, Oligodendrocytes, T cells, Metabolically active glioblastoma cells, and Mesenchymal glioblastoma cells. The dot plot (Fig. 9B) delineates the expression profiles of various genes across different cell types, with particular emphasis on the expression of ELN, TGFBR1, SSTR2, FCER1G, CDK4 and BIRC5 genes in multiple cellular populations. The t-SNE visualization (Fig. 9C) elucidates the expression distribution of the target genes at the single-cell level, revealing their differential expression patterns across various cell types.
Fig. 9
Single-cell expression analysis. A t-SNE diagram; B Lattice diagram; C t-SNE map of gene expression distribution
Single-cell communication analysisIn Fig. 10A, the intercellular signalling network diagram reveals complex signal transduction relationships among various cell types, such as metabolically active glioblastoma cells, mesenchymal glioblastoma cells, and inhibitory neurons, with node size representing cell types and connection thickness indicating signal strength. Figure 10B shows a heatmap of outgoing signalling patterns across different cell types, where the x-axis represents signalling pathways, the y-axis represents cell types, and colors indicate signal intensity, highlighting the diversity of intercellular communication. Figure 10C displays a heatmap of the relative strength of different signalling pathways in various cell types, with the x-axis representing signalling pathways, the y-axis representing cell types, and colors indicating signal intensity, illustrating the specificity of signal transduction in different cell types. Finally, Fig. 10D depicts the PTN signalling pathway network diagram, where nodes represent cell types and connection thickness indicates the strength of PTN signal transduction, emphasizing the critical role of PTN signalling in intercellular communication. These results collectively reveal the complexity of intercellular communication networks and the specificity and intensity of signalling pathways among different cell types.
Fig. 10
Single-cell communication analysis. A Intercellular signaling network diagram; B Heatmap showing outgoing signaling patterns across different cell types; C Heatmap displaying the relative strength of different signaling pathways in various cell types; D PTN signaling pathway network diagram
Clinical relevance of model genesFor subsequent basic research, clinical diagnostic and prognostic analysis of individual model genes are conducted. To determine the diagnostic value of model genes for gliomas, receiver operating characteristic (ROC) curves are respectively plotted, and the results show that the predictive efficacy of each model gene is very good (Fig. 11A–C). To determine the predictive value of model genes for the survival status of gliomas, survival curves (Kaplan-Meier curves) are respectively plotted and presented in forest plot format, and the results show that the survival predictive efficacy of each model gene is very good (Fig. 11D).
Fig. 11
The predictive and diagnostic efficacy of model genes in glioma. A–C ROC curves of model genes; D Forest plot of Kaplan-Meier curves of model gene
Model gene immunoinfiltration relevanceClinical diagnosis and prognostic analysis of individual model genes are conducted to determine the role of model genes in the immune infiltration of gliomas. The results show that all model genes are associated with a variety of immune cells (Fig. 12A), participating in diverse immune cell functions (Fig. 12B), and also influencing comprehensive immune scores, stem cell scores, and tumor heterogeneity scores (Fig. 12C).
Fig. 12
The immune infiltration relevance of the model gene in glioma
IKBKE promotion on gl261 cell migration and apoptosisThe study examined the impact of IKBKE overexpression and underexpression on GL261 cell migration and apoptosis. The findings demonstrated that the downregulation of IKBKE in GL261 cells notably suppressed both cell migration (Fig. 13A–C) and apoptosis abilities (Fig. 13D–G).
Fig. 13
IKBKE promotion on GL261 Cell Migration and Apoptosis. A–C Compared to the control group, siRNA-IKBKE inhibits cell migration between the three groups, D compared to the control group, siRNA-IKBKE promoted cell apoptosis, E the grayscale bands of Bcl-2 and Bax, F and G the statistical bar graphs of Bcl-2 and Bax
留言 (0)