
記住我

Randomized controlled trials are widely considered the gold standard for assessing the efficacy of new treatments. By design, they minimize confounding as a challenge to internal validity. However, participants enrolled in trials are often not representative of the patient populations that will receive treatment in clinical practice, limiting the external validity of trial results.1,2 When treatment effects vary by factors related to trial enrollment, estimated treatment effects from trials may not directly extend to clinical practice.3,4
Increasingly, researchers are interested in quantitatively extending observed treatment effects from trials to target populations to improve population-level decision-making. The aim of such analyses is to optimize both internal and external validity, together termed target validity.5 Methods for extending (via generalizing or transporting) treatment effects to target populations are under rapid development in epidemiology and statistics.4–16 These methods require, among other steps,17 (1) identification of a target population; and (2) identification, measurement, and specification of at least one variable set which is sufficient for achieving conditional exchangeability between the trial population (sometimes called the study sample) and the target population, thereby enabling valid treatment effect estimation in the target population.
To date, identifying variable sets for extending treatment effects has often focused on including variables hypothesized to: (1) have distributions that differ between the trial and target populations and (2) modify the effects of treatment on the outcome on the scale of interest–i.e., effect measure modifiers.3 While theoretical (i.e., directed acyclic graphs [DAGs])18,19 and statistical20 guidance for variable selection approaches are developing, data visualization –may offer accessible guidance on, and clarify the process of, variable selection, particularly for applied practitioners.
We introduce a practical roadmap for extending estimates from trials to clinical practice settings that uses four visualizations to inform variable selection for models to extend treatment effects and assess model specification and performance. We illustrate this roadmap using a landmark phase III randomized controlled trial, the Multicenter International Study of Oxaliplatin/5-Fluorouracil/Leucovorin in the Adjuvant Treatment of Colon Cancer (MOSAIC)21 and a target population of patients diagnosed with stage II or III colon cancer identified within a network of community oncology practices in the United States.22
METHODS Conceptual FoundationsEffect measure modification is present when the effect measure (e.g., risk ratio) for a given exposure–outcome relationship varies across levels of another covariate.23 As noted, the identification of variable sets for extending treatment effects focuses on the inclusion of variables that: (1) have distributions that differ between the trial and target populations and (2) are effect measure modifiers. Notably, these criteria are marginal: they describe whether marginal distributions of a variable (e.g., income) differ between trial and target populations and whether there is an effect measure modification by this variable without considering levels of other variables that explain either discrepancy (e.g., age). However, when choosing variables for confounding control and internal validity,24 selecting variables for addressing external validity requires consideration of conditional, rather than marginal associations.18
Theorized conditional associations between potential effect measure modifiers, trial participation, and the outcome can be elucidated using selection diagrams11,12 or causal DAGs,5–8,11,12,17,18,25 hereafter both referred to as DAGs. DAGs can be used to identify theoretical variable sets of effect measure modifiers for extending treatment effects to target populations.18 However, in practice, a theoretically important effect measure modifier (e.g., based on a DAG) may not be practically important for extending treatment effects for a specific trial and target dataset. Consequently, variable selection approaches using data in hand are often needed, exactly as with control of confounding, where we might identify a confounder using a DAG which is of little practical importance in a particular analysis, and so we might choose to not control for this confounder for statistical efficiency.
Existing Methods and Visualization InputsSelecting variable sets of effect measure modifiers for treatment effect transport from trials to clinical practice often starts with model specification using DAGs. Once a DAG has been defined, one or more statistical models must be specified. Current methodologic approaches for extending treatment effects from trials to target populations require either (1) defining a statistical model for the outcome(s) given treatments and other covariates (so as to implement a g-computation approach),26 (2) a statistical model of how patients are “sampled” into the trial and target populations given treatments and other covariates (so as to implement an inverse weights or odds of sampling weighting approach),4 or (3) both (doubly robust approaches).27
Below, we describe two statistical models that will be used as inputs for the proposed visualizations. Note that these visualizations can be useful to researchers regardless of the methodologic approach selected for extending treatment effects.
Input model #1—Outcome model (using the trial data only). The purpose of this outcome model is to establish whether specific covariates of interest might be effect measure modifiers on any scale (e.g., risk difference, risk ratio), which, in a randomized trial setting, would require that they be associated with the outcome, conditional on other covariates. Notably, this approach differs from other approaches28–31 which seek to identify potential effect measure modifiers on a specific scale through estimation of treatment effects within subgroups (i.e., heterogeneous treatment effect estimation). Specifically, we rely on the observation that predictors of an outcome will be effect measure modifiers on some scale,32 assuming there is an effect of treatment on the outcome (in at least some subgroup).33
For our motivating study, we include all trial participants (treated and untreated), assume a binary outcome (e.g., 5-year all-cause mortality), and model the log odds of the outcome as a function of a set of potential effect measure modifying variables, Zk, in Equation 1:
ln[Pr(outcomei=1|Z)Pr(outcomei=0|Z)]=β0+β1Z1+…+βkZk.
Because we are using trial data alone, neither treatment nor interaction terms are included in Equation 1 because randomization has, in expectation, already removed any causal paths that flow from covariates to the outcome through treatment.
Input model #2—Trial “sampling” model (using both the trial and target population data). The purpose of this model is to identify whether any potential effect measure modifiers are also conditionally associated with “sampling” into the trial versus a target population. To estimate these model parameters, researchers can concatenate (or “stack”) the individual-level trial and target data. This concatenated dataset must contain the same potential effect measure modifiers from both the trial and target data. Alternative approaches can be applied.3,34
In our motivating example, we model the log odds of being in the trial as a function of the specified variables, Zk, in Equation 2:
ln[Pr(triali=1|Z)Pr(triali=0|Z)]=β0+β1Z1+…+βkZk.
Using the parameter estimates from the sampling model above (Equation 2), researchers can compute the “probability” of trial “sampling” for each trial participant as a function of their own covariates, Zk. We put “probability” and “sampling” in quotes (but not hereafter) because these parameters reflect study design rather than true population parameters. However, they can be treated as true population probabilities for purposes of transport.16 Next, we calculate stabilized inverse odds of sampling weights in Equation 3.4
Wi=Pr(triali=0|Zi)Pr(triali=1|Zi)×Pr(triali=1)Pr(triali=0)
Each trial participant is assigned a weight, wi, based on the probability of trial sampling. All individuals in the target population are assigned a weight of 0 and their information is not used further.
Overview of RoadmapBuilding upon this foundation, we introduce a practical roadmap (Figure 1) to guide applied researchers in extending treatment effects from trials to clinical practice using sampling weights as an analytic approach.
 FIGURE 1.:
FIGURE 1.: Practical roadmap to guide the extension of treatment effects from a trial to a target population. Tasks and suggested approaches/visualizations are stated at each step of the roadmap, noting that steps 3–6 may require iteration.
Step 1 is to identify variables that may be conditionally associated with the outcome, and thus potential effect measure modifiers on at least one scale, based on existing knowledge via literature review, clinical input, and DAGs.18 This approach will necessarily be conservative, as some of these variables may not be required for valid transport on the scale of interest.19
Step 2 is to screen these potential effect modifiers using the data at hand. For this step, we introduce the Variable Importance for Treatment Transport plot. This plot displays (1) conditional relationships between potential effect measure modifiers and the outcome in the trial population only (i.e., Equation 1) and (2) conditional relationships between potential effect measure modifiers and selection into the trial versus target population (i.e., Equation 2). In our motivating example, we approximate both sets of relationships via logistic regression models with linear and additive parameterizations. This approximation allows a simple assessment of whether, in the study data, a variable appears to be associated with the outcome and trial sampling once other variables have been conditioned upon. On the x axis, this figure shows the conditional association between patient characteristics and trial participation (vs. being in the target population) by estimating adjusted odds ratios (aORs). On the y axis, the figure shows conditional associations between the same patient characteristics and a binary study outcome, displaying estimated aORs, using the trial data only. To reflect uncertainty from the estimation of the aORs, we apply bootstrap resampling using 1000 replicates of both the trial and target data to generate 95% confidence interval (CI) “clouds” for these associations.
Step 3 uses the potential effect measure modifiers from step 2 to build a more flexible sampling model that can include nonlinearities and interaction terms. This more fully specified sampling model will be used to generate inverse odds of sampling weights.
Step 4 takes the model refined in step 3 and uses it to evaluate the assumption of external positivity.17 When the probabilities estimated from the true trial sampling model (the sampling probability, given combined strata of all covariates) are >0 for covariate strata represented by all individuals in the target population, we can say that the analysis meets the external positivity assumption.15 However, we note that it is possible to have empirical violations of external positivity if patterns of potential effect measure modifiers observed in the target population do not exist in the trial population. In this setting, we should consider whether extrapolation beyond or interpolation within data is reasonable. For visualization, we adapt a visualization of estimated propensity score distributions. Similar to Stuart et al.,35 we compare the distribution of probabilities from the sampling model in the trial and target populations.
Step 5 is then to evaluate the balance of the effect measure modifiers included in the sampling model before and after weighting. We use a diagnostic to assess whether the model balances potential effect measure modifiers (identified from the plot in step 2) across the trial and target populations. This is an adaptation of the Love plot used for assessment of internal validity visualizing covariate balance across treatment groups before and after propensity score weighting. In our adaptation, we visually compare distributions of the proposed effect measure modifiers between the trial and target population. For each covariate, we use standardized mean differences to quantify differences across populations as:
Standardizedmeandifference=M1−M2SDpooled,
where M1 is the mean value in the target, M2 is the mean value in the trial population, and SDpooled is the pooled standard deviation. This equation can be modified to accommodate continuous and categorical variables.16 We then plot these standardized mean differences before and after weighting to visually assess improvements in variable balance across the trial and target population. Any residual imbalances could signal the need to enrich the trial sampling model (e.g., by returning to step 3 and adding interaction terms or splines). This type of iterative model building to achieve covariate balance has been described previously.15
Step 6 performs a diagnostic evaluation of potentially influential observations with large weights. We visualize the distribution of estimated sampling weights in the trial population. This graphical depiction overlays a vertical line at the percentile of the weighted trial population where the weights exceed 10, a commonly used threshold for defining influence.36 This visualization can also be stratified according to subgroups (e.g., cancer substage) that may highlight potential drivers of large weights. These plots could be used with quantitative summary measures (i.e., mean weights and upper and lower percentiles) to justify sensitivity analyses applying weight truncation or trimming to reflect a less variable target population.10,37
Step 7 involves reviewing, synthesizing, and evaluating output from steps 2–6 (possibly in an iterative fashion) to finalize a variable set of effect measure modifiers required for extending treatment effects from a trial to a target population.
Data Sources and Study PopulationsWe apply this roadmap to a trial comparing two adjuvant chemotherapies for colon cancer and a target population of patients treated within community oncology practices.
Trial DataData from the MOSAIC trial were accessed through Vivli.org,38 a data-sharing platform facilitating access to trial data. The MOSAIC trial evaluated the efficacy of combination 5-fluorouracil (5FU) plus oxaliplatin (FOLFOX) compared with 5FU alone in reducing mortality for stage II/III colon cancer.21 Key eligibility criteria included age 18–75 years, resected stage II or III colon cancer, Eastern Cooperative Oncology Group performance score (a measure of physical function) of two or lower,39 and no prior chemotherapy or radiotherapy. Patients were enrolled from 1998 to 2001 across 146 centers in 20 countries. Food and Drug Administration approval for stage III colon cancer was based on 3-year disease-free survival showing the benefit of FOLFOX versus 5FU (hazard ratio [HR] = 0.78; 95% CI = 0.65, 0.93).21 Benefits were attenuated in stage II (HR = 0.84; 95% CI = 0.62, 1.14) and thus this indication was excluded from the Food and Drug Administration label, although FOLFOX is used off-label.40
Target Population DataThe target population for a given study should be the relevant population for whom we want to generate new evidence about the effects of a specific treatment decision. In oncology care, new therapies tend to quickly disseminate into clinical practice, which is the case for FOLFOX.40 In this dynamic environment, identifying clinically relevant target populations can be challenging. For example, following a new cancer drug approval, individuals who remain on the “old” standard of care often represent a population who lack the indication for or who have a contraindication against (e.g., high risk of peripheral neuropathy) the “new” treatment. Thus, in such settings, researchers may consider limiting the target population to individuals who receive the “new” treatment only, as the treatment decision following drug approval is not relevant for those who receive the “old” treatment.
To identify our target population for this study, we used the US Oncology iKnowMed electronic health record database,41 an oncology-specific, web-based, electronic health record system capturing outpatient data for patients treated by >1000 physicians at >400 community oncology practices from 2008 to 2019. Each year, iKnowMed captures ~10% of newly diagnosed cancers in the United States. We obtained patient-level data on all stage II and III colon cancer patients in iKnowMed from 1 January 2008 to 30 June 2019.
We first identified those who met MOSAIC eligibility criteria. Applying eligibility criteria to the target population helps to address external positivity. To identify a clinically relevant target population in a contemporary, postapproval period, we included those patients initiating FOLFOX only. The FOLFOX-treated population represents patients selected by oncologists who were likely to benefit from treatment, considering the risk of their tumor progressing, the potential toxicities of treatment, and their overall life expectancy.
Potential Effect Measure ModifiersIn each data source, we measured the following variables theorized via clinical input to be potential effect measure modifiers of the effect of FOLFOX versus 5FU alone on 5-year mortality: age, sex (female or male), cancer stage (II or III) and substage (IIA, IIB/C, IIIA, IIIB, and IIIC), performance status (0, 1, or 2), and body mass index (BMI, continuous). For the Variable Importance for Treatment Transport plot, we modeled age as a continuous variable (scaled to 20-year increments) and all other variables were included as binary variables. For all subsequent visualizations, the trial sampling model allowed more flexibility including age as a cubic restricted spline with three knots, and more flexible categories for all other variables (e.g., 0 vs. 1 vs. 2, <18.5 BMI vs. 18.5–25 BMI vs. 25–30 BMI vs. 30+ BMI, stage IIA or IIB vs. IIC vs. IIIA vs. IIIB vs. IIIC).
Statistical AnalysisWe estimated the average treatment effect in the MOSAIC trial population by computing the HR for all-cause mortality after 6 years from trial start comparing FOLFOX with 5FU using Cox proportional hazards regression. Using the final variable set selected by applying the roadmap, we then extended the average treatment effect from the trial to our target population by estimating inverse odds of sampling-weighted HRs for all-cause mortality comparing FOLFOX and 5FU. We computed 95% CIs using bootstrapping with 10,000 replicates, resampling both the MOSAIC and target populations.
This study was considered exempt by our Institutional Review Board. We used SAS version 9.4 (Cary, NC) and R (4.0.3) for analyses.
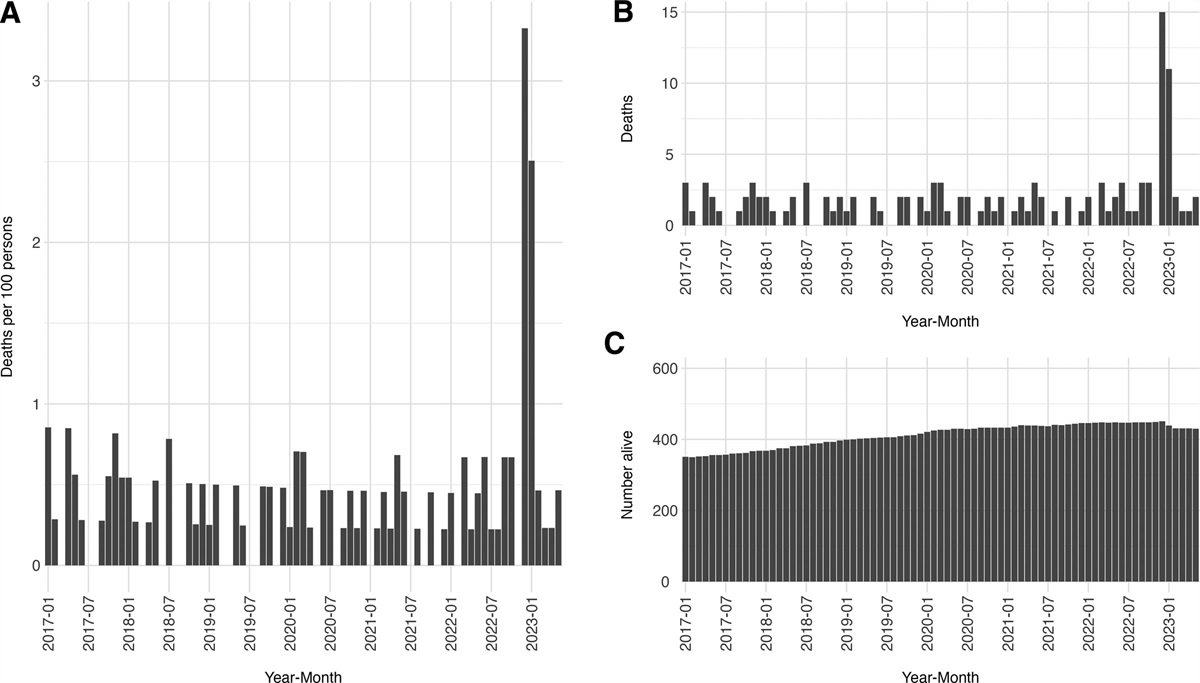
RESULTSThere were 2246 participants enrolled in MOSAIC and 12,486 stage II or III colon cancer patients identified iKnowMed; 3266 initiated FOLFOX and met eligibility (83% of the original FOLFOX-treated in iKnowMed, eFigure 1; https://links.lww.com/EDE/C88).
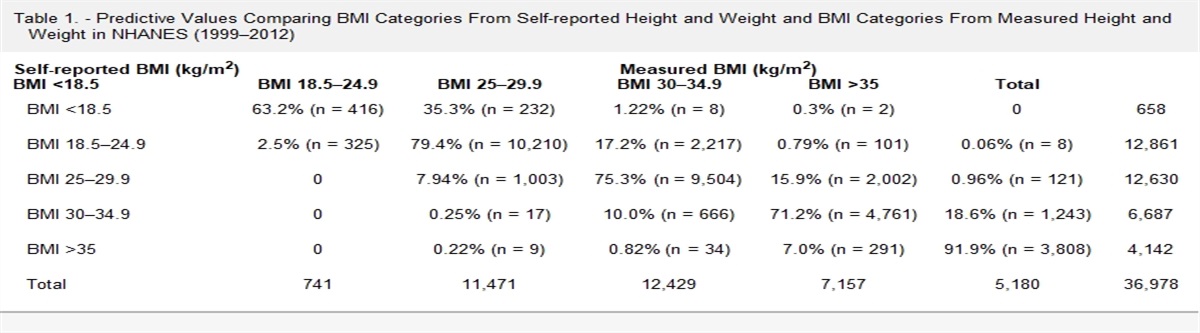
The Table reports the distribution of characteristics from the trial and target populations. Age distributions were similar between the two populations. Given the MOSAIC trial was largely conducted in Western European countries, racial composition differed between populations. The distribution of cancer substage and body mass index also varied substantially.
TABLE. - Description of MOSAIC Trial and US Oncology Target Populations MOSAIC Trial US Oncology Target Variable Overall (N = 2246) FOLFOX-Treated (N = 3266) Age group (N, %) 18–25 9 (0%) 11 (0%) 26–35 59 (3%) 94 (3%) 36–45 189 (8%) 294 (9%) 46–55 494 (22%) 832 (26%) 56–65 805 (36%) 1075 (33%) 66–75 690 (31%) 960 (29%) Sex (N, %) Male 1218 (54%) 1688 (53%) Female 1028 (46%) 1578 (48%) Race (N, %) White 2188 (97%) 2221 (68%) Other 58 (3%) 433 (13%) Not documented – 612 (19%) Year of diagnosis (N, %) 2008–2010 – 790 (24%) 2011–2013 – 852 (26%) 2014–2016 – 886 (27%) 2017–2019 – 738 (23%) Cancer substage (N, %) IIA 728 (32%) 248 (8%) IIB or IIC 171 (8%) 237 (7%) IIIA 100 (4%) 430 (13%) IIIB 786 (35%) 1755 (54%) IIIC 461 (21%) 595 (18%) Tumor differentiation (N, %) Undifferentiated – 17 (0%) Poor 290 (13%) 572 (18%) Moderate 1414 (63%) 1699 (52%) Well 434 (19%) 614 (19%) Documented unknown 108 (5%) 23 (1%) Not documented – 341 (10%) Performance status (N, %) 0 675 (30%) 1354 (41%) 1 1277 (57%) 1821 (56%) 2 294 (13%) 91 (3%) CEA test (yes vs. no) 2159 (96%) 2910 (89%) BMI category <18.5 80 (4%) 72 (2%) 18.5–<25 1130 (50%) 994 (31%) 25–<30 761 (34%) 1126 (35%) 30+ 275 (12%) 1037 (32%) Not available – 37BMI indicates body mass index; CEA indicates carcinoembryonic antigen.
Figure 2 shows the Variable Importance for Treatment Transport plot comparing the MOSAIC and target populations (step 2 on the roadmap). The multivariable aORs for each potential effect measure modifier and MOSAIC trial participation are plotted on the x axis and the multivariable aORs for 5-year all-cause mortality are plotted on the y axis. The color clouds represent results generated in bootstrap resamples.
 FIGURE 2.:
FIGURE 2.: Variable importance for treatment transport plot used to inform variable selection for transport of MOSAIC trial to the US Oncology target population. This figure shows the multivariable odds ratios (ORs) for each baseline patient characteristic and MOSAIC trial enrollment (x axis) and 5-year all-cause mortality (y axis). The point estimate for each variable is surrounded by a 95% confidence interval cloud, estimated using 1000 bootstrap replicates. Each color represents a different baseline variable: red is age (in 20-year increments), gray is sex (female vs. male), blue is BMI (30+ vs. <30), yellow is performance status (0–1 [good function] vs. 2 [worse function]), and green is cancer stage (II vs. III disease).
The green cloud at the bottom of the figure represents cancer stage (II vs. III), which is strongly associated with lower 5-year all-cause mortality and higher odds of trial participation (i.e., there was a higher proportion of stage II patients in the trial than in the target). The gray cloud centered on the x axis shows that sex is not independently associated with 5-year all-cause mortality. Based on this finding, we dropped sex from the sampling model. Our final model included age, performance status, BMI, and stage.
Figure 3 provides a visualization to check whether the probability of trial sampling is >0 for all individuals in the target population (step 4 on the roadmap). The mean probabilities of trial sampling in each population (vertical lines) showed large differences. Importantly, however, there were no individuals in the target population where the probability for trial sampling fell outside of the range of trial population probabilities (i.e., no evidence of empirical violations of external positivity).
 FIGURE 3.:
FIGURE 3.: Distribution of predicted probabilities for trial sampling in the MOSAIC population versus the US Oncology target population. The dark gray bars represent probability distributions for the MOSAIC trial population and the light gray bars represent probability distribution for the US Oncology target populations. The dashed vertical lines represent the mean probabilities for each population. These graphs provide a composite view of the similarity of the trial and target populations with respect to the proposed set of effect measure modifiers (i.e., those variables included in the trial sampling models). In addition, they serve as a visual means to assess empirical violations of external positivity (i.e., areas of nonoverlap in the histograms) and further consideration of issues of extrapolation and interpolation.
Figure 4 displays the standardized mean differences for baseline variables comparing the target population with the MOSAIC population (step 5 on the roadmap). The overlayed vertical lines denote a threshold of ±10%, which is often used by researchers as meaningful to determine covariate balance.42 Candidate variables showing residual imbalances after weighting might indicate poor trial sampling model specification. Before inverse odds of sampling weighting, we see that performance status, BMI, and cancer substage have large imbalances. After weighting, all variables appear well-balanced for the target population.
 FIGURE 4.:
FIGURE 4.: Love plots reporting the standardized mean differences comparing baseline patient characteristics in the MOSAIC trial and US Oncology target population. The MOSAIC trial population serves as the referent population. Standardized mean differences for each baseline variable for the FOLFOX-treated target population are shown. The circles show the standardized mean differences before inverse odds of sampling weighting and the diamonds show these after weighting. The vertical lines are placed at −10% and 10%, as commonly used benchmarks for covariate balance in propensity score matched or weighted analyses.
Figure 5 displays the distribution of inverse odds of sampling weights for the weighted MOSAIC trial population (step 6 on the roadmap). This plot aims to identify potentially influential observations. We define extreme weights as stabilized weights that surpass 10 (one individual has 10 times more influence than they did before weighting). Overall, these plots show there are few extreme weights in the target population. If influential weights were more prevalent, sensitivity analyses using truncation or trimming may be warranted.10 Similar plots further stratified by cancer stage and BMI category (underweight, normal, overweight, obese) are included in eFigure 2; https://links.lww.com/EDE/C88.
 FIGURE 5.:
FIGURE 5.: Distribution of stabilized inverse odds of sampling weights applied to the MOSAIC trial population for transport to the US Oncology target population. A horizontal line at 10 denotes a threshold commonly used to identify potentially influential observations; the vertical line denotes the point in the distribution at which the weights exceed 10.
Extending the MOSAIC Trial Results to the Target PopulationIn the MOSAIC trial at 6 years,43 overall survival was 78.5% in the FOLFOX arm and 76% in the 5FU arm, with a HR of 0.84 (95% CI = 0.71, 1.00). When we used the final set of effect measure modifiers and model specification noted above, we estimated an inverse odds of sampling-weighted HR of 0.94 (0.73, 1.19), showing attenuation.
DISCUSSIONThere is increasing interest in evaluating the external validity of treatment effects from trials to inform population-level health decisions.44,45 Quantitative methods to extend treatment effects from trials to target populations are promising, but like any inferential approach require both causal and statistical assumptions. The four visualizations proposed here provide accessible means for evaluating several of these assumptions.
One assumption is that of conditional exchangeability, or that sampling into the trial should not be associated with the outcome conditional on covariates. The Variable Importance for Treatment Transport plots introduced here, with background knowledge and clinical input, can help identify a potentially smaller set of variables required for conditional exchangeability given the data in hand, acknowledging that we can only transport effects based on patterns observed in the trial population. A second assumption for valid transport requires external positivity where the predicted conditional probability of trial sampling must be >0 for all individuals in the target population. Overlap of estimated probabilities from the trial sampling model provides evidence that this assumption is reasonable, given the data, and lack of overlap may indicate bias. A third assumption is that of correct model specification. To check this assumption, we plotted the standardized mean differences after inverse odds of sampling weighting. All variables appear well-balanced within the target population, which provides necessary, but not sufficient evidence of correct model specification. In the future, researchers may consider applying nonparametric machine learning algorithms to relax this assumption. However, the performance of these approaches is only guaranteed under large sample sizes that may not be available in applied settings. Simulations could be used to establish finite sample performance in specific settings. The assumptions addressed by our visualizations are neither sufficient nor exhaustive, though other assumptions, such as consistency, are not amenable to simple graphics and may be best addressed via subject matter knowledge.
Methods for extending treatment effects from trials to target populations usually require researchers to specify either (1) outcome models conditional on covariates or (2) sampling models conditional on covariates (or both models). Regardless of the approach, Variable Importance for Treatment Transport and Love plots can help assess the feasibility of transporting trial results to specific target populations and prioritize variable selection. Inverse odds of sampling weight distribution plots require explicit calculation of weights via a sampling model, but this diagnostic remains useful for outcome-modeling approaches to highlight if inference relies on a small proportion of individuals.
We prioritized simplicity of our approach and vis
留言 (0)