
記住我
In a recent study led by Dr. Hisashi Arase published in Cell, a new mechanism underlying the breakdown of self-tolerance in systemic lupus erythematosus was reported that autoreactive T cells recognize neoself-antigens. Neoself-antigens, encompassing a range of self-molecules, were shown to be presented on major histocompatibility complex class II by non-canonical mechanisms following the downregulation of the invariant chain protein.
Systemic lupus erythematosus (SLE) is a prototypical systemic autoimmune disease marked by the breakdown of immune tolerance. The presence of autoantibodies, particularly antinuclear antibodies, serves as a diagnostic hallmark of SLE. These autoantibodies are produced by autoreactive B cells with necessary help from autoreactive T cells. In a normal immune system, multiple checkpoints maintain immune tolerance to self-antigens.1 Hence, autoimmunity often arises from failures in central tolerance which prevents the development of autoreactive lymphocytes, or peripheral tolerance which inhibits the activation or differentiation of mature autoreactive lymphocytes.
Studies using various animal autoimmunity models have revealed that dysregulation of diverse pathways can compromise key checkpoints in central and peripheral tolerance. These pathways include failure of autoreactive T cells to undergo clonal deletion in the thymus, defective regulatory T (Treg) cell generation and function, excessive differentiation of follicular helper T (Tfh) cells and peripheral helper T (Tph) cells, defective free DNA clearance, and exaggerated activation and survival of self-reactive B cells2,3 (Fig. 1). The heterogeneity in SLE aetiology is also thought to contribute to its diverse clinical manifestations.
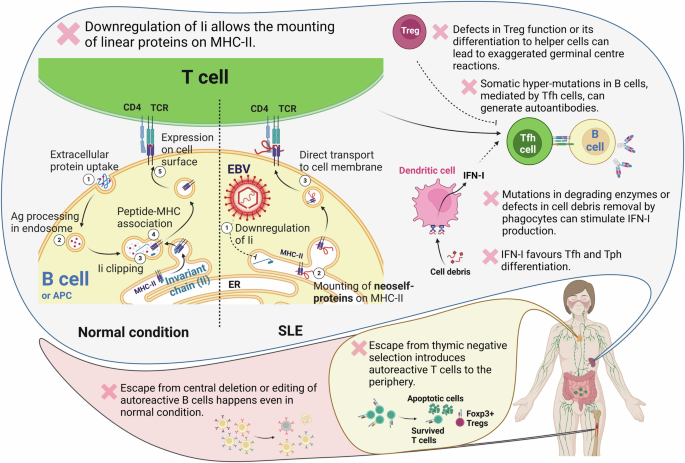
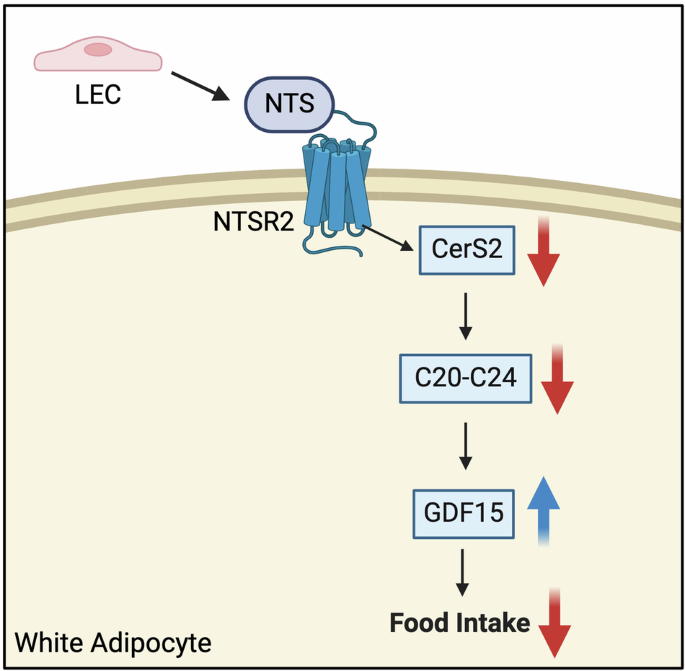
Fig. 1: Defective expression of Ii promotes neoself-antigen presentation via MHC-II, leading to lupus-like disease.
Ii regulates MHC-II migration to the cell membrane. Under physiological conditions, Ii is processed and removed from MHC-II once it merges with endosomal vesicles containing degraded proteins. This allows appropriate peptide presentation on MHC-II, facilitating its transport to the cell surface. However, if Ii expression is disrupted (for instance due to EBV reactivation), self-proteins from the ER can aberrantly bind to MHC-II. This neoself-antigen–MHC-II complex is directly transported to the membrane, leading to the activation of autoreactive Tfh cells and the initiation of autoimmunity. This novel role of Ii complements other immune tolerance checkpoints including clonal deletion of autoreactive T and B cells, development and function of Treg cells, effective clearance of extracellular DNA, and regulation of proinflammatory cytokine IFN-I.
To answer the question of why self-molecules are recognized as non-self, post-translational modifications, such as citrullination, are well-studied in rheumatoid arthritis (RA), where patients with anti-citrullinated protein antibodies show a specific phenotype characterized by exaggerated inflammatory lung pathology.4 However, the underlying molecular mechanisms in SLE is still unclear. During antigen presentation, invariant chain (Ii) binds to major histocompatibility complex class II (MHC-II) in the endoplasmic reticulum (ER) and plays dual roles by transporting MHC-II to endosomes and lysosomes and preventing spontaneous binding to proteins.5 The potential involvement of Ii in autoimmune diseases was first proposed in the 1990s.6 Over a decade ago, Dr. Hisashi Arase’s group demonstrated that, in the absence of Ii, linear proteins can bind to the MHC-II groove and be transported to the cell surface, bypassing the endosomal compartment.5 Therefore, they are not processed into peptides that are typically presented during thymic T cell development and selection.
To investigate the impact of Ii regulation on the immune system in vivo, the authors employed a tamoxifen-inducible Ii-knockout (Ii-iKO) mouse model. They showed that deleting Ii in adult mice induced a lupus-like disease, marked by increased Tfh cells, germinal centre and switched memory B cells, and plasma cells,7 indicating hyperactivation of Tfh cell-driven autoimmune responses.8 Other observed phenotypes included altered spleen size, body weight, and survival curves. Interestingly, unlike the Ii-iKO mice, germline Ii-KO mice did not develop autoreactive antibodies, suggesting thymic negative selection of corresponding T cell clones.1
To further characterize the specificity and function of lymphocytes in the autoimmune-prone Ii-iKO model, the authors conducted single-cell RNA sequencing (scRNA-seq) with TCR/BCR profiling. In Ii-iKO mice, there was clonal expansion of effector B cells, including plasma cells. Testing recombinant antibodies with expanded BCR sequences from Ii-iKO mice revealed their binding to cell nuclei and cytoplasm, indicating autoantibody production. Parallel analyses of T cells in this model revealed that ~20% of TCRs from Ii-iKO mice were autoreactive to Ii-iKO splenocytes and promoted autoantibody production in Ii-KO mice, suggesting that autoreactive Tfh cells are present in this mouse model.7
The authors also explored whether Ii influences autoreactivity in human T cells. They selected one SLE patient and engineered CD4+ T cells with expanded TCRs based on scRNA/TCR-seq data, and then compared these T cells in response to cells with matched HLA-DR in the presence or absence of Ii expression. Eight out of 85 engineered T cell clones exhibited significantly higher responses to Ii-deficient cells than to Ii-sufficient cells, indicating that ~10% of expanded T cell clones in this SLE patient showed autoreactivity to neoself-antigens caused by Ii deficiency. Notably, these eight TCRs were primarily found in T cell clusters identified as Treg and Tfh cells, suggesting a mix of effector and regulatory autoreactive functions. Furthermore, only two of these TCRs showed > 10-fold increase in in vitro TCR reporter assays when comparing Ii-sufficient and Ii-deficient antigen presentation, while the others displayed ~2-fold differences between the two conditions.7 This suggests that Ii deficiency partially influences T cell activation in this human subject.
The Ii-iKO mouse model and the analysis of human T cell clones’ response to Ii-deficient cells provide convincing evidence supporting the notion that reduced Ii expression can significantly enhance the presentation of unaltered self-antigens through MHC-II, thereby driving the expansion of neoself-antigen-specific T cells and promoting autoimmunity. However, the remaining question is why Ii expression is downregulated in SLE. The authors demonstrated that Epstein-Barr virus (EBV) reactivation could contribute to this effect, at least partially, through the overexpression of BZLF1. Epidemiological and serological studies have shown a positive association between EBV infection and autoimmune diseases, including SLE, Sjögren’s syndrome, and multiple sclerosis.9 Further research is needed to explore the role of EBV infection in Ii downregulation and to determine whether this regulation extends to other autoimmune diseases. Additionally, given the critical role of cytokines, such as interferon type I (IFN-I), in the aetiology of SLE and their reported impact on antigen presentation via immunopeptidomic studies,10 their potential influence on Ii expression should be carefully examined.
Overall, the discovery of Ii downregulation, as a molecular mechanism for immune tolerance breakdown, suggest Ii expression as a possible checkpoint in preventing the generation of autoreactive Tfh cells and restricting autoimmunity (Fig. 1). The broader significance of this mechanism in human SLE development and its potential generalization to other autoimmune diseases require investigation across larger cohorts. In particular, quantifying the frequency of Ii deficiency-dependent autoreactive T cells among the total T cell population will be essential. Such insights will aid the precise management of SLE as one of the most heterogeneous autoimmune diseases.
留言 (0)