RDD is a rare non–Langerhans cell histiocytosis, having a reported prevalence of 1:200,000, and it mostly occurs in young individuals [2]. The exact etiology of RDD is unknown and probably multifactorial. It may be associated with autoimmune diseases (lupus, idiopathic juvenile arthritis, autoimmune hemolytic anemia) [4], tumors (Hodgkin’s lymphoma and non-Hodgkin’s lymphoma) and infections such as varicella-zoster virus, human herpesvirus 6 [5], Epstein–Barr virus (EBV), cytomegalovirus [4], Parvovirus [6], Klebsiella [7], and Brucella [8], as well as genetic factors (reported germ-line mutations in SLC29A3 in familial cases) [4]. In this case report, we describe a patient with a history of ankylosing spondylitis. To date, very few reports in the literature have described RDD in conjunction with a history of ankylosing spondylitis. The combination of ankylosing spondylitis with lymphadenopathy (sinus histiocytosis) was first reported in 1986 [9]. In 2006, a hospital in Taiwan reported a patient with both RDD and ankylosing spondylitis [10]. As an autoimmune disease, the potential association between ankylosing spondylitis and RDD remains unclear. We report what may be the first case of intrathoracic RDD in a patient with concurrent ankylosing spondylitis, highlighting this rare but valuable case that provides relevant evidence for further investigation into the relationship between RDD and immune-mediated diseases such as ankylosing spondylitis.
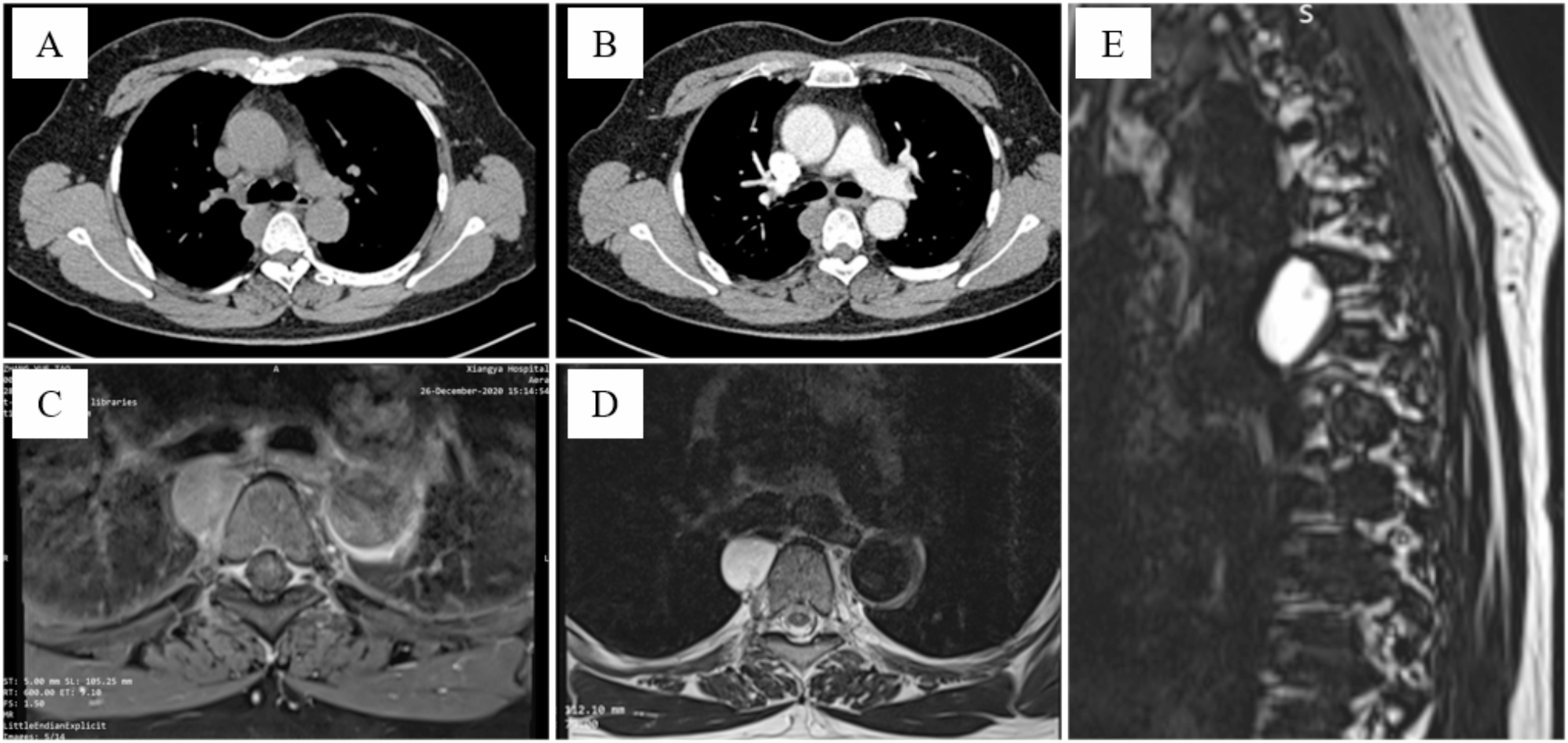
The diagnosis of RDD can be particularly challenging. Patients diagnosed with RDD may exhibit clinical manifestations including fever, night sweats, and weight loss. Additionally, they may present with non-specific laboratory abnormalities such as anemia, leukocytosis with neutrophilia, and polyclonal hypergammaglobulinemia [3, 4]. In lymph node cases, CT shows enlarged nodes with uniform enhancement. Intrathoracic RDD appears as nodular consolidation in lung lobes, possibly with pleural effusion, fibrosis, or nodules [1]. On T1-weighted MRI, affected lymph nodes resemble muscles in signal intensity, while on T2-weighted MRI they appear hyperintense with uniform enhancement. 18 F-FDG PET/CT shows increased metabolic activity [11]. Due to the lack of specific laboratory and radiological findings, early diagnosis of RDD is notably formidable.
Isolated thymic RDD is exceedingly uncommon, as the majority of intrathoracic RDD cases manifest as pulmonary or peritracheal masses and pleural lesions [12, 13]. RDD typically exhibits inert growth, with only rare cases showing invasive growth. Mediastinal RDD represents an exceedingly small fraction, comprising just 0.5% or less of all lesions occupying the mediastinal space [14]. Currently reported anterior mediastinal RDDs are mostly isolated mediastinal masses, with less vascular invasion [15, 16]. RDD in the thymus shares similarities in appearance, FDG avidity, and distribution with several other diseases. It poses a wide range of potential differential diagnoses, including thymoma, lymphoma, metastatic disease and proliferative inflammatory diseases like inflammatory myofibroblastic tumor [12]. Zhang et al. [16] also reported a case of RDD presenting as an anterior superior mediastinal mass. Initially, they diagnosed it as thymoma, but the final pathological result confirmed it to be a mediastinal RDD with KRAS mutation. In our clinical practice, huge anterior mediastinal masses are often invasive, especially thymomas, which typically show vascular infiltration on enhanced CT and increased FDG uptake on PET-CT [11]. In this case, the patient did not show typical clinical symptoms of RDD or abnormal results from preoperative laboratory tests. Two previous mediastinal biopsies did not provide conclusive evidence for a diagnosis, posing a challenge to diagnosis. Due to the patient’s young age, we proposed a mediastinal biopsy under thoracoscopy, but the patient refused. Considering the typical signs of malignancy and vascular invasion in the anterior mediastinal mass, we eventually opted for surgical treatment.
Diagnosis of RDD requires sufficient tissue samples, and biopsies should be reviewed by a pathologist familiar with RDD [3].Histologic examination of nodal RDD reveals sinus expansion by large histiocytes, with abundant pale or “watery-clear” cytoplasm (a large hypochromatic nucleus with a prominent nucleolus [3]. Extranodal lesions exhibit more fibrosis, fewer RDD histiocytes, and less emperipolesis. The immunophenotype of RDD histiocytes includes positivity for S100 and fascin, variable positivity for CD68, CD163, CD11c, and CD14, and negativity for CD1a and CD207. Plasma cells express markers such as CD38, CD138, MUM1, and IgG4 [1,2,3,4]. BCL-2 and OCT2 are commonly expressed in RDD cases [17, 18]. As primary thymic RDD is extremely rare, clinicians and radiologists often find it difficult to make a definite diagnosis without definitive pathological findings. RDD exhibits typical cytological features, making fine needle aspiration cytology (FNAC) a commonly used diagnostic tool for RDD. However, diagnosing extranodal RDD through FNAC may be more challenging. In our case, despite two attempts at needle biopsy, the limitations of the FNAC meant that we did not obtain definitive evidence of RDD in the early stages of treatment. Its nonspecific clinical presentation, subtle variations, atypical features, and rarity may lead cytopathologists to overlook or misinterpret the condition. In contrast to the limited tissue samples obtained through FNAC, thoracoscopic biopsy yields tissue specimens that are more likely to preserve adequate tissue architecture. Through direct visual inspection and multi-angle sampling, it offers more comprehensive and diverse diagnostic information. Therefore, we consider that for young patients with mediastinal masses difficult to diagnose conclusively via FNAC, further refinement with thoracoscopy or mediastinoscopy biopsy may be a more appropriate diagnostic step.
Treatment for RDD varies depending on individual clinical circumstances. While spontaneous resolution occurs in 50% of cases, about 10% may face complications leading to death [1]. Asymptomatic patients can be monitored, and surgical resection is an option for those with specific symptoms or solitary extranodal lesions. Systemic treatments such as glucocorticoids, sirolimus, radiotherapy, chemotherapy, and immunomodulatory therapy may be viable options for patients with multifocal or unresectable lesions [1, 4, 19]. Immunotherapy with TNF-α inhibitors, thalidomide, and lenalidomide have also been attempted due to elevated levels of TNF-α and interleukin-6 in RDD patients [4, 19]. For inoperable and refractory RDD, treatment with thalidomide may be an exciting option. The report by Hoyo-Muñoz et al. [20] suggests that thalidomide therapy may induce remission in patients who have failed fourth-line systemic chemotherapy. If relevant driver mutations are identified, targeted therapies like tyrosine kinase inhibitor(imatinib) [21], BRAF inhibitor (dabrafenib) [22], and MEK inhibitor (trametinib) [23] have shown some efficacy in treating refractory RDD patients. Overall, a multidisciplinary approach is recommended for optimal outcomes.





留言 (0)