
記住我


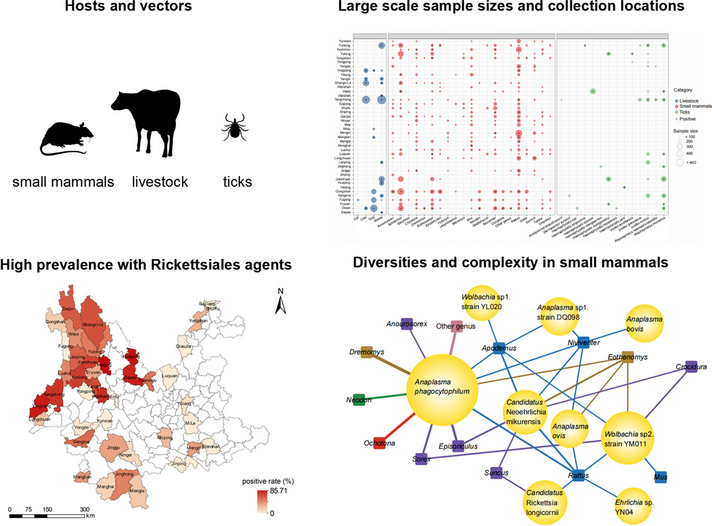

Between May 18, 2011 and November 23, 2020, a total of 7964 organisms were collected from 42 investigation counties (Table 1). Among them, 4330 small mammals were identified, representing 57 species across 21 genera, 10 families, and 4 orders. Rattus tanezumi emerged as the dominant species with the highest constituent ratio (720/4330, 16.63%) and exhibited the broadest distribution across 28 counties (Fig. 1, Table S2). Livestock samples comprised of 2375 individuals, representing 4 species, with goat (Capra aegagrus) as the predominant species (446/2375, 18.78%). Of the 1259 ticks collected, 829 were parasitized ticks and 430 were host-seeking ones, classified into 14 species under 5 genera of Ixodidae. Rhipicephalus microplus ranked as the dominant species (457/1259, 36.30%), followed by Ixodes ovatus (302/1259, 23.99%) (Fig. 1, Table S2).
Table 1 Overall prevalence of animals and vectors from different counties with emerging rickettsiae in Yunnan ProvinceFig. 1
Distribution of different species of samples and positive samples in different counties in Yunnan Province. Blue, red and green circles represent samples of livestock, small mammals and ticks, “ + ” represent sites, hosts and vectors with positive results for rickettsiae. The circles size of symbols represents the number of samples
Ten species under five genera emerging Rickettsiales in small mammalsIn total, 2.59% (112/4330) of sampled individuals and 24 out of 57 (42.11%) species of small mammals were infected with rickettsial agents, primarily concentrated in western counties of Yunnan Province. The highest prevalence of rickettsial agents was observed in Yuanmou County (11.06%), followed by Weixi County (10.00%), with significant differences (P < 0.001) (Table 1). Among the 21 identified genera, the genus Rattus exhibited the highest frequencies of rickettsial agent infections (n = 51), with R. tanezumi having a prevalence of 3.06%. The genus Dremomys had the highest prevalence (5.60%, 1/18). Significant differences in prevalence were observed among genera of small mammals (P = 0.025) (Fig. 2A). Furthermore, the highest prevalence of rickettsial agents was observed in areas with altitudes above 4000 m (P = 0.004) (Fig. 2B). Additionally, the prevalence of rickettsiae was higher in males than in females (P = 0.004). Small animals collected from alpine meadows exhibited the highest prevalence, followed by those in mixed forests (P = 0.046) (Fig. 2C).
Fig. 2
Comparison the positivity rate of rickettsiae in different altitudes, hosts, and habitats in Yunnan Province. A Host genus (P = 0.025) B Altitude (P = 0.004) C Habitat types (P = 0.046)
To better understand the potential transmission of rickettsial agents in small mammals, we constructed host-rickettsial correlation networks. Among the small mammals studied, 21 genera were found in 10 families. Of these, 14 genera in 6 families of small animals were found to be harboring rickettsial agents. Some rickettsial agents were specifically found to infect only one genus, such as Wolbachia sp. strain YL020 in the genus Apodemus, Anaplasma bovis in the genus Niniventer, and Ehrlichia sp. YN04 in the genus Rattus. Conversely, the genera Neodon, Dremomys, Ochatona, and Anourosorex were found to carry A. phagocytophilum, and the genus Mus was detected to host only Wolbachia sp. strain YM011, indicating host tropism, a preference towards certain rickettsia. However, both A. phagocytophilum and Candidatus Neoehrlichia mikurensis (CNM) were detected in more than 7 genera of small animals (Fig. 3), indicating a broad host spectrum.
Fig. 3
Host-rickettsial correlation network topology. Rectangles represent the genera of the small mammals; the size of the circle represents the level of rickettsial agents’ positivity and the thickness of the straight line represents the level of host infection with rickettsiae
As revealed in Fig. 4, 10 different species of rickettsial agents were detected. Among these, A. phagocytophilum, A. ovis, CNM, and Rickettsia typhi were confirmed as human pathogens. CNM exhibited the highest positivity rate (1.04%, 45/4330), followed by A. phagocytophilum (0.97%, 42/4330) in small mammals. Notably, the sequences of samples DQ098 and DQ292 were identical and revealed the closest similarity (97.65%) to an uncultured Anaplasma sp. clone T7 (GenBank No. KU189193). This was provisionally termed Anaplasma sp. strain DQ098. Additionally, YL-020 and the other eight identical sequences represented by YM-011 had the highest similarities of 99.68% with OX366385 and 98.72% with CP116767, respectively, representing two distinct Wolbachia spp. (Table S3).
Fig. 4
Phylogenetic tree based on partial sequences of 16S rRNA (660 bp) gene of the rickettsiae species derived from small mammals. Phylogenetic analysis was performed using the neighbor-joining method and trees were tested by bootstrapping (1000 replicates)
The constructed phylogenetic tree revealed that the rickettsial species detected in the small mammals clustered into five major groups: Neoehrlichia, Ehrlichia, Anaplasma, Wolbachia, and Rickettsia (Fig. 4). Within the Wolbachia cluster, the sequences identified were significantly different from known species and were categorized into two clades, termed Wolbachia sp. strain YL020 and Wolbachia sp. strain YM011.
High prevalence of eight species emerging Rickettsiales in livestocksThe positivity rate in livestock blood samples collected from 16 counties was 33.68% (800/2375). The highest prevalence was observed in Yingjiang County (81.65%, 89/109), followed by Huaping County (66.02%, 68/103), with significant differences (P < 0.001). Among the four types of livestock sampled, Bovine taurus exhibited the highest number of infected individuals and the highest prevalence of rickettsial agent infections (45.03%, 317/704, P < 0.001). The phylogenetic tree indicated that the rickettsial strains detected in livestock clustered into three distinct groups: Ehrlichia, Anaplasma, and Wolbachia. The predominant species identified were A. ovis, A. marginale, and A. phagocytophilum. Notably, A. phagocytophilum displayed considerable diversity, with at least 3 variants differing by 1 to 30 base pairs, though they were located within the same clade in the phylogenetic tree (Fig. 5).
Fig. 5
Phylogenetic tree based on partial sequences of 16S rRNA (660 bp) gene of the rickettsiae species derived from livestock. Phylogenetic analysis was performed using the neighbor-joining method and trees were tested by bootstrapping (1000 replicates)
High prevalence and wide distribution of Rickettsiales in ticksThe overall positivity rate among 1259 ticks, encompassing 14 species in 5 genera, was 20.65% (260/1259). Rickettsiales agents were observed in all 14 sampling counties, with infestations of 8 out of 14 (57.14%) tick species. The genus Rhipicephalus had the highest prevalence (37.07%, 175/472, P < 0.001). Specifically, the prevalence of Rickettsiales in the dominant tick species R. microplus and Haemaphysalis montgomeryi was 36.54% (167/457) and 28.86% (58/201), respectively. H. yeni exhibited the highest prevalence (85.71%, 12/14, P < 0.001). A total of 13 species of rickettsiae were detected in the tick samples, of which two were identified as pathogenic to humans (Fig. 5). The main tick-associated rickettsial agents were Candidatus R. longicornii (n = 147) and A. ovis (n = 67). Among the 14 sampling sites, Jianchuan County had the highest number of positive ticks (n = 126), while Heqing County had the highest prevalence (85.71%, 12/14, P < 0.001), reflecting their different population sizes. Additionally, ticks collected at altitudes of 2000 to 2499 m had the highest prevalence of rickettsiae (27.08%, 88/325, P = 0.004). The prevalence of parasitic ticks (28.47%, 236/829) was significantly higher than that of host-seeking ticks (5.58%, 24/430, P < 0.001). Notably, ticks parasitizing Bos taurus exhibited the highest prevalence (53.84%, 49/91, P < 0.001).
BLAST analysis revealed that the 16S rRNA of HQ-T-1 had 98.49% identity with uncultured Anaplasma sp. clone D9_6 (GenBank No. MK814441), JC-T-222 had complete identity with uncultured Anaplasma sp. clone Dedessa (GenBank No. KY924886), and TC-T-400 had 99% identity with OL690561, forming a distinct branch in the phylogenetic tree. Combined with phylogenetic analyses, HQ-T-1, JC-T-222, and TC-T-400 were confirmed as monophyletic groups, named Anaplasma sp. strain HQT1, Anaplasma sp. strain JCT222, and Anaplasma sp. strain TCT400, respectively (Fig. 6).
Fig. 6
Phylogenetic tree based on partial sequences of 16S rRNA (660 bp) gene of the rickettsiae species derived from ticks. Phylogenetic analysis was performed using the neighbor-joining method and trees were tested by bootstrapping (1000 replicates)
Characterization of seven pathogenic rickettsial and Candidatus Rickettsia longicornii with multigene analysisSeven tick samples (WX-T-85 to WX-T-91) positive for Candidatus R. longicornii from Weixi County were selected for sequencing of the entire ompA, gltA, ompB, 17 kDa, 16S rRNA (nearly full length), sca4, and sca1 genes (Fig. 7A-G). The ompA, ompB, 16S rRNA, and sca1 genes displayed 100% homology with the sequences of Candidatus R. longicornii or Rickettsia endosymbiont of Haemaphysalis longicornis. The gltA and 17 kDa genes from WX-T-90 and WX-T-91 had 100% homology with the sequences of Candidatus R. jingxinensis (GenBank Nos. MW114883 and MW114879). The sca4 gene sequence revealed 99.51% homology with Candidatus R. longicornii (GenBank Nos. MK620855, MG906677). Despite some conflicting sequences, the rickettsial organism is likely Candidatus R. longicornii.
Fig. 7
Phylogenetic trees constructed by the MEGA v.6.0 software based on the neighbor-joining method of multigene detected in ticks infected by Candidatus Rickettsia longicornii. A ompA gene (384 bp); B gltA gene (381 bp); C ompB gene (400 bp); D 17 kDa gene (395 bp); E 16S rRNA gene (nearly full length); F sca4 gene (843 bp); G sca1 gene (429 bp)
Both the full-length 16S rRNA gene (Fig. S1A) and the groEL gene of CNM (Fig. S1B) detected in this study were remarkably distinct from other sequences detected in China (Heilongjiang), Germany, and Switzerland. Among the three clusters of CNM based on the groEL gene the phylogenetic tree indicated that the present sequences belong to cluster I (southwest) and cluster III (southeast) [37] (Fig. S1B). Although the sequences from Lushui and Weixi were close to the southwest branch (I), they clustered into small branches, indicating they differ from previous ones in Yunnan.
Five Anaplasma-associated pathogens (A. capra, A. ovis, A. bovis, A. marginale, and A. phagocytophilum) were confirmed in samples. The groEL (Fig. S2B) and gltA (Fig. S2C) genes of A. capra revealed 100% homology between Capra aegagrus in Tengchong and patients in Heilongjiang [8]. The msp4 gene (Fig. S2D) of A. capra had 99.8% homology, indicating pronounced similarity with the pathogen infecting patients. Variations of A. capra were also detected in Tengchong, Deqin, and Shangri-La (Fig. S2). The phylogenetic tree based on the msp4 and entire 16S rRNA gene (Fig. S3A) of A. ovis formed a cohesive branch closely aligned with reference sequences. Sequences detected in Yunlong consistently clustered within a separate branch, demonstrating a close genetic relationship across different hosts within this geographic region. The gltA gene (Fig. S3C) of A. ovis distributed into two small branches. The groEL gene of A. bovis, A. marginale, and A. phagocytophilum also indicated diversity and distinctive distribution among their host ticks and animals (Fig. S4). The groEL and gltA genes of E. canis clustered in a distinct branch along with other sequences identified in ticks in China, with sister branches from Thailand, the Philippines, Spain, and France. The Dsb and TRP36 genes of E. canis formed unique, singular clades (Fig. S5).
留言 (0)