
記住我

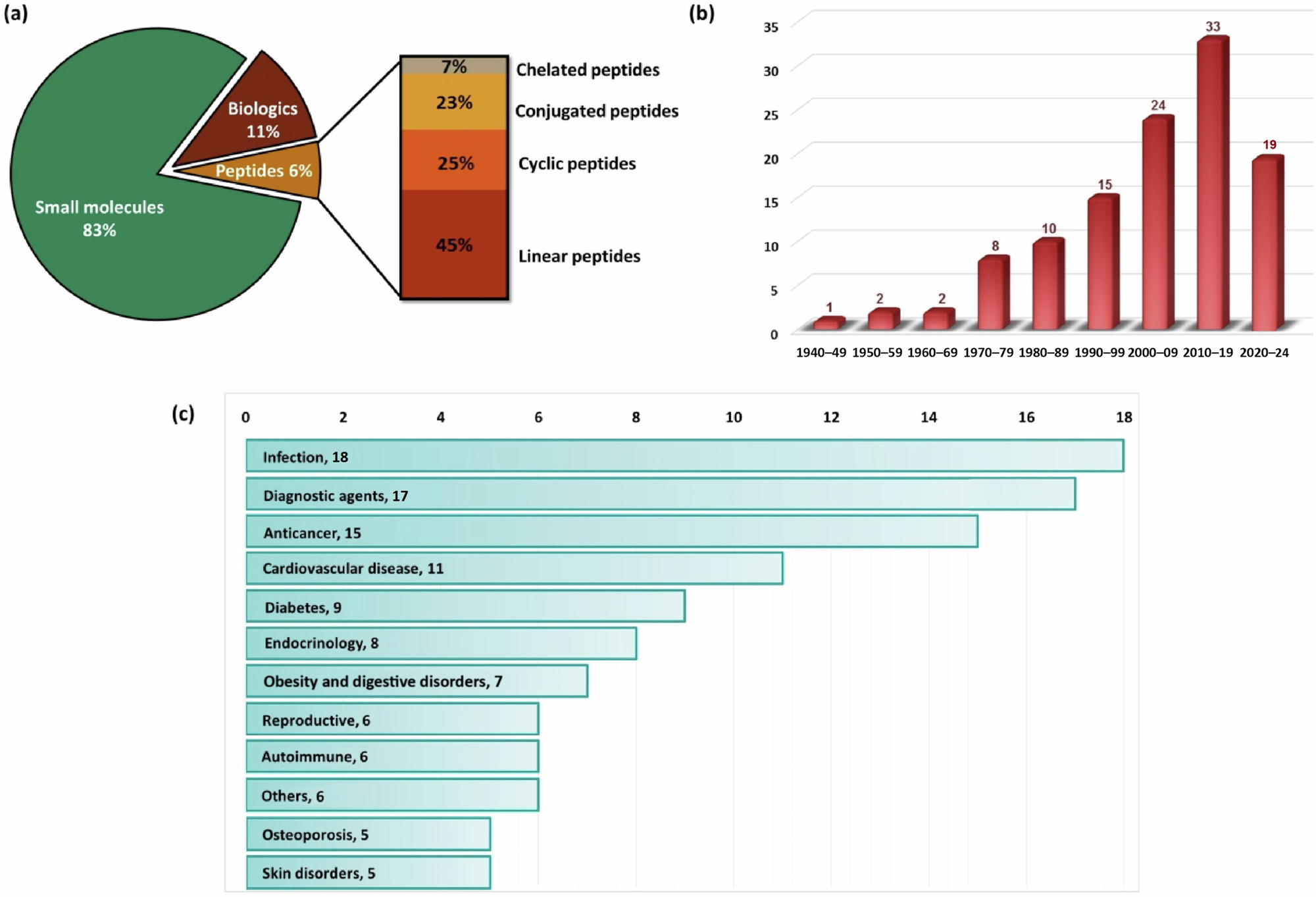
Given this special issue honors the scientific contributions of Dr. Ronald Borchardt, we considered fitting to briefly discuss one of the areas where his research has made a great difference: strategies to improve the stability and activity of protein- and peptide-based drugs. Due to their natural origin and vast similarity with endogenous molecules, therapeutic peptides, which are made of defined amino acid sequences typically ranging from 500 to 5000 Da, represent a distinct and distinguished class of medicinal agents. Since the isolation of the 51-amino acid long peptide hormone insulin in 1921 (which may be considered the first therapeutic peptide), significant advancements have been made in the area resulting on the approval of over 110 peptide-based drugs worldwide until May 2024 (Fig. 1). With an estimated value of US$39.3 billion in the worldwide market and a projected annual growth rate of 6.4%, peptides are a substantial component of the pharmaceutical industry. Further, it is estimated the market share of peptide-based drugs will increase to US$68.7 billion by the year 2030 [1].
Fig. 1
a Classification of the US Food and Drug Administration (FDA) approved molecules with subdivisions of peptides on the basis of structures approved from 1940 until May 2024; b decade-wise distribution of peptide-based drugs approved by the FDA; c distribution of peptide drugs according to each application. Adapted and reprinted with permission from reference [1]. Copyright Elsevier 2023
Peptide drugs can mimic hormones, growth factors, neurotransmitters, or ion channels and its ligands. They can also work as anticancer and anti-infective agents [2]. A chief example of a peptide drug is enfuvirtide, a 36-amino acid biomimetic peptide used to treat human immunodeficiency virus (HIV). This peptide works by preventing the fusion of the virus with the target human cell. Another example is the incretin mimetic liraglutide. This molecule is an analog of the human glucagon-like peptide 1 (GLP-1) and increases insulin release from the pancreas. The peptide consists of a palmitic acid tail (in blue, Fig. 2) grafted to glutamic acid (in red) that is connected to a lysine residue (in green). The peptide acts as a therapy against Type 2 diabetes mellitus or chronic obesity. Thus, peptides have emerged as a key area of focus in pharmaceutical research that has advanced in recent decades due to progress in structural biology, recombinant biologics, new synthetic and analytical technologies. This progress has greatly sped up peptide drug development, leading to the establishment of a sophisticated system that encompasses peptide drug discovery, design, synthesis, structural modification, and biological evaluation.
Fig. 2
Chemical structure of liraglutide highlighting the lysine residue (green) attached to the glutamic acid linker (red) and palmitic tail (blue)
Peptide-based drugs exert their mechanism of action by altering a myriad of biological functions. For example, they can interact with cell surface receptors or with an intramolecular target, they may alter protein-protein interactions or damage cell membranes. Peptides induce intracellular effects with high specificity and affinity, sharing a similar mechanism of action with biologics, i.e., therapeutic proteins, and antibodies (due to hydrogen bonds, salt bridges, and Van der Waals interactions) or can exert their action through electrostatic interactions as in the case of membrane-disrupting antibacterials. A key advantage is that therapeutic peptides exhibit lower immunogenicity and reduced production costs compared to biologics.
Unfortunately, peptide-based drugs may possess some limitations such as the inability to permeate cell membranes, low bioavailability, and non-specific interactions with plasma proteins. Further, unlike proteins, natural peptides lack the added structural stability provided by secondary or tertiary structures. Therefore, peptides may lose their secondary structure, and consequently, their activity. Also, the amide bonds that hold the amino acids together in peptides are susceptible to hydrolysis or enzymatic degradation when exposed to biological environments. These inherent chemical properties render peptides chemically and physically unstable, resulting in a short half-life and rapid elimination from the body. Such weaknesses pose an obstacle in the development of peptide drugs.
As expected, the biological activity of a peptide is a direct result of its chemical structure. Thus, several strategies have been developed to stabilize secondary structure or to improve metabolic stability to maintain, or enhance, their biological activity. Further chemical (structural) changes can be implemented to achieve better selectivity or solubility. Prior to modifying the structure of a primary peptide drug candidate, it is crucial to identify the minimum active sequence with the desired biological properties. In other words, to find the elements that are essential for activity and the residues that can be replaced without compromising biological action. The classical sequence scanning method, known as alanine-scanning [3], is commonly utilized to substitute each residue with alanine, generating a series of lead peptide analogs. This approach helps in determining which specific residues are responsible for the biological activity of the lead peptide: a decrease in activity indicates the importance of the replaced residue while a minimal change in activity suggests that the replaced residue was dispensable. Subsequent modifications of the replaceable residues, as well as the C- and N-termini of the lead peptide, are then conducted to produce the desired compound.
Among the possible peptide alterations, backbone modification serves as a crucial approach to enhance the proteolytic stability of peptides. The identification of proteolytic sites within the peptide can be achieved through stability studies and metabolite determination. Various methods of backbone modification exist, such as the substitution of L-amino acids with D-amino acids, the insertion of N-methylated amino acids, and the incorporation of β-amino acids and peptoids. By introducing such residues into the peptide sequence, particularly at sites prone to proteolysis, the plasma half-life of peptide drugs can be effectively prolonged. Analogs of natural amino acids like homoarginine, benzyltyrosine, and β-phenylalanine (Fig. 3) are readily accessible and can be effectively utilized for the chemical modification of peptide side chains during synthesis. Various GLP-1 analog drugs, including liraglutide and semaglutide, have undergone side chain modifications [4].
Fig. 3
Chemical structures of benzyltyrosine (a), β-phenylalanine (b) and homoarginine (c)
The insufficient strength of weak forces in peptides, including hydrogen bonds, Van der Waals forces, and intramolecular hydrophobic interactions, may not allow for a stable secondary structure conformation. To create potential peptide drug candidates, it is necessary to make further adjustments to the backbone, N- or C-termini, or side chains to replicate the structures found in natural products or key areas in protein-protein interactions (PPI) and enhance the stability of secondary structures. One of the most common strategies to achieve enhanced stability is cyclization, which can be divided in different types of connections including head-to-tail, backbone-to-side chain, and side chain-to-side chain cyclization. Dr. Borchardt’s group has reported some strategies for the preparation of cyclic prodrugs of peptides by linking the N-terminal amino group to the C-terminal carboxyl group of peptides using an acyloxyalkoxy group [5]. The cyclization strategy offers several advantages besides stabilizing the secondary structure of the peptide including increased proteolytic stability and cell-permeability. When a single peptide sequence is not connected to other peptides, it cannot form loops or turn structures. However, cyclization plays a crucial role in facilitating the formation of these secondary structures by organizing intramolecular interactions [6]. Moreover, peptide cyclization is frequently employed to stabilize other secondary structures like α-helices and β-sheets. The α-helix, which is predominantly formed by intramolecular hydrogen bonds, makes up 90% of helical structures. Imitating the α-helix in peptides facilitates the discovery of regulators of PPIs [7].
The stability of the α-helix can be enhanced by creating cross-links via side chains or by substituting hydrogen bonds with covalent bonds, known as hydrogen bond surrogates (HBS) (Fig. 4). In the α-helix configuration, the side chains of amino acids at positions i, i + 4, and i + 7 are located on the same side and establishing cross-links between i and i + 4 or i and i + 7 effectively brings together backbone atoms, aiding in the formation of hydrogen bonds within helical structures. Various types of cross-links have been explored, including lactam-based cross-links, which involve the creation of a lactam bridge between the side chain of glutamic acid or aspartic acid and lysine, the formation of disulfide bonds by substituting residues with cysteine or homocysteine and the use of bis-electrophilic linkers [8]. Stapled peptides, another cross-linking technique, have been developed to stabilize the α-helix structure by replacing natural amino acids with non-natural ones at the i and i + 4 or i and i + 7 positions, forming bonds with cross-links [9]. The HBS modification approach entails replacing a hydrogen bond of the α-helix peptide with a covalent bond to pre-arrange the helical structure. Another category of protein secondary structures is represented by β-sheets and β-strands, which are formed using turn mimics. Another common strategy to favor α-helices consists in using 2-aminoisobutyric acid (Aib) or di-n-propylglycine (Dpg). These modifications should increase metabolic stability due to the presence of unnatural amino acids and to enhance α-helix formation without affecting biological activity [10]. To enhance the stability of β-sheets, peptides can be modified by incorporating D-amino acids, such as D-Pro, to create a turn structure within the sequence. D-Pro-L-Pro templates are widely recognized as effective frameworks for stabilizing antiparallel β-hairpins in various PPI inhibitors. Additionally, the creation of β-sheets and β-strand structures has been achieved through techniques like macrocyclization or the application of amyloid β-sheet mimics [11].
Fig. 4
Strategies for peptide cyclization and stabilization of α-helices, β-sheets and β-strands. Adapted and reprinted with permission from reference [19]. Copyright 2022 Springer Nature Limited
The discussion above centered on increasing the structural stability by linking segments of by tuning stability using unnatural amino acids. However, another strategy used to prolong the lifespan of peptide and protein therapeutics is through the attachment of polymers. The most common one is PEG, which consists of repetitive units of ethylene oxide, a polymer that is non-biodegradable, non-toxic, and has low immunogenicity [12]. By undergoing PEGylation, the effective molecular weight of proteins can be increased, thereby reducing their elimination through renal clearance via kidney filtration. Additionally, the presence of the PEG moiety can protect proteins from degradation by proteolytic enzymes through steric hindrance and enhance their absorption by improving water solubility. These advantageous properties have made PEGylation a widely utilized approach for modifying therapeutic proteins, and it has been successfully employed since the 1970s to optimize protein therapeutics.
Lipids and larger proteins are commonly attached to enhance the pharmacokinetics of peptide. Well-known peptide drugs like liraglutide, semaglutide, and insulin degludec have been linked with C14/16/18 fatty acids, resulting in prolonged plasma circulation and decreased degradation during renal excretion. Additionally, serum albumin and immunoglobulin are utilized to extend the circulation time of peptides by increasing their molecular weight beyond the glomerular filtration cut-off. This approach has been employed to increase the half-life of dulaglutide and albiglutide, which are administered through weekly injections [13].
Peptide modifications have proven to be effective in enhancing the activity and stability of peptides, making them more suitable for use as drugs. However, peptides are susceptible to degradation by digestive enzymes in the stomach and intestine, which limits their oral administration. Indeed, the number of orally administered peptide-based drugs approved thus far is limited to just 11 [14]. Thus, it is imperative to explore novel strategies in order to augment this count. To overcome this challenge, researchers have explored various routes for delivering them. One promising approach is co-formulation with permeation enhancers, which enables the oral administration of peptide drugs. For example, semaglutide has been conjugated with C18 fatty acid and approved for once-weekly subcutaneous injection [15]. This formulation exhibits greater plasma stability compared to other GLP-1 analogs. Furthermore, semaglutide has been co-formulated with sodium N-[8-(2-hydroxybenzoyl) amino]caprylate (SNAC) for oral administration in the treatment of Type 2 diabetes mellitus. The inclusion of SNAC in the formulation prevents the degradation of semaglutide in the stomach by reducing the activity of digestive enzymes. Additionally, the hydrophobic nature of SNAC enhances the lipophilicity of semaglutide, facilitating its absorption through the gastric membrane and its subsequent transport into the systemic circulation. Other formulation strategies to improve peptide stability can be mentioned including self-emulsifying drug delivery systems (SEDDS), solid lipid nanoparticles (SLN), liposomes and micelle nanoformulations, and microgels. Furthermore, researchers are exploring alternative delivery methods such as pulmonary administration, transdermal delivery, and the use of implantable pumps for specific peptide drugs [16]. Notably, the development of inhalable insulin, peptide vaccine [17], neuropeptides [18], and micro-implantable pumps for insulin delivery are areas of active investigation. It is anticipated that these technologies will be increasingly utilized for the delivery of a wider range of peptide drugs in the future.
In summary, peptides have emerged as a distinct category of therapeutic agents in recent years due to their unique biochemical properties and therapeutic potential. Despite outperforming small molecules and large biologics in certain aspects, peptides often face challenges such as poor membrane permeability and in vivo stability attributed to the inherent limitations of amino acids. Extensive research efforts have been dedicated to the exploration, manufacturing, and enhancement of peptide drugs to address these limitations. The fusion of conventional lead peptide discovery techniques with innovative methodologies like rational design offers a reliable strategy for the rapid development of effective lead compounds. These peptides can be further tailored in a site-specific manner through chemical synthesis to boost their stability and efficacy. Significant advancements in molecular biology, peptide chemistry, and peptide delivery technologies have promoted progress in peptide drug discovery, production, and therapeutic applications. Given the significant therapeutic potentials, market prospects, and economic values associated with therapeutic peptides, it is anticipated that they will continue to increase investment and research attention, ultimately leading to sustained success in the long term.
留言 (0)