Protein–protein interactions (PPIs) play an important role in cellular biochemical events, regulating biological information and directing cell destiny. Understanding the pathogenic process of human illnesses is aided by research into the involvement of PPIs. HSPs can interact with numerous proteins to conduct a number of biological roles in heart disease, such as autophagy, inflammation, endoplasmic reticulum stress, and oxidative stress.
3.1. HSP60 and Atrial FibrillationHSP60 is a mitochondrial protein that also exists in the cytosol, endoplasmic reticulum, and plasma membrane. The function of HSP60 is mostly determined by its distribution. HSP60 and HSP10 are required proteins for the proper function and homeostasis of mitochondrial proteins. HSP60 regulates protein folding and prevents protein aggregation in the mitochondria. HSP10, a mitochondrial chaperone, aids in the folding and assembly of proteins transported into the mitochondrial matrix [
19]. In cardiomyocytes, high levels of HSP60 and HSP10 exert protective effects on electron transport chain (ETC) complexes. Complexes III and IV are specifically elevated in cardiomyocytes overexpressing HSP60 or HSP60/10. In mice, HSP60 deletion leads to decreased mitochondrial complex activity, decreased mitochondrial membrane potential (ΔΨmito), and increased reactive oxygen species (ROS) generation [
20]. Additionally, intracellular overexpression of HSP60 protects cardiac myocytes from apoptosis by blunting the release of cytochrome c and the activation of caspase-3 [
19]. This antiapoptotic effect of HSP60 may be in part because HSP60 binds to Bax (B-cell lymphoma-2 associated X) and prevents its translocation into mitochondria, whereas the complex is destroyed under hypoxia, resulting in mitochondrial dysfunction [
21,
22]. Instead of atrial myocytes, ventricular cardiomyocytes or hypoxia-reoxygenation cardiac myocyte models (hypoxia for 6 hours in an anaerobic workstation) were utilized in these studies. The precise mechanism by which HSP60 protects against AF is unknown. As fully discussed in
Section 4.1 below, the researchers use the HSP60/10 characteristics to predict the onset of AF and the stabilization of sinus rhythm after undergoing mitral valve surgery.Conversely, extracellular HSP60, as a ligand of Toll-like receptor 4 (TLR4), induces myocyte apoptosis through the TLR4–MYD88–p38/nuclear factor kappa-B (NF-κB) pathway, which may increase AF burden [
23]. Both tachypaced atrial HL-1 cardiomyocytes and left atrial appendages (LAAs) of paroxysmal or persistent AF patients display mitochondrial stress, as evidenced by increased HSP60 and HSP10 expression, decreased ATP production, a loss of the mitochondrial membrane potential, and mitochondrial network fragmentation, resulting in contractile dysfunction and AF progression [
24]. 3.2. HSP70 Family and Atrial Fibrillation
The HSP70 family (HSP70s) (size range: 70−78 kDa) is classified into two groups: constitutive members (HSC70, HSP75, HSPA5 (glucose-regulated protein 78 [GRP78] and HSPA9 [GRP75])) and inducible members (HSP70), all of which are ATP dependent. HSP70s are expressed in different cellular locations, including the cytosol, nucleus, endoplasmic reticulum (ER), and mitochondria, and are secreted. Their main functions are to maintain the dynamic balance of the synthesis, folding, degradation, and translocation of proteins.
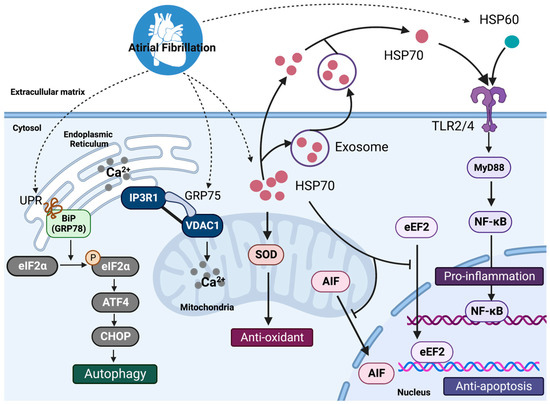
Recent research has revealed that ER-stress (ERS) and the subsequent activation of macroautophagy (hereafter autophagy) play an important role in the onset and maintenance of AF [
25,
26]. The ERS-induced activation of autophagy is an essential mechanism of atrial remodeling in AF. Autophagy is an evolutionarily conserved protein-breakdown mechanism that sequesters damaged or expired proteins and organelles into autophagosomes for eventual lysosomal degradation. While autophagy is required for normal physiological function, excessive autophagy is harmful. Brundel, B.J. and colleagues developed experimental cardiomyocyte and dog models that mimic persistent AF in patients and exhibit reversible electrical and irreversible structural remodeling. HL-1 atrial cardiomyocytes were treated with 6 Hz tachypacing (1 Hz for normal pacing) to induce a frequency rise with tachypacing (six-fold increase) comparable to what happens during human AF. Tachypacing-induced contractile dysfunction of HL-1 cardiomyocytes, and the ER-stress response, can activate the autophagy–lysosome pathway via the unfolded protein response (UPR) [
26]. The UPR stimulates the phosphorylation of the α-subunit of eukaryotic initiation factor 2 (eIF2) at serine 31, which inhibits protein translation and initiates the selective expression of stress-responsive genes such as activating transcription factors (ATF) 4 and 6. ATF4 and ATF6 signaling, in turn, increase the expression and activation of the CCAAT/enhancer-binding protein (C/EBP) homology protein (also known as CHOP) and various autophagy proteins, such as autophagy related 12 (ATG12), MAP1LC3B (also known as LC3) and BiP (also known as GRP78), which together stimulate autophagosome elongation and autophagic protein degradation [
14,
27]. ER-stress inhibition with the pharmacological chaperone 4-phenylbutyrate (4-PBA), overexpression of the ER chaperone GRP78, or mutation of the eIF2 gene could limit autophagy and thus prevent electrical and contractile failure in AF models [
26]. The aforementioned results were also confirmed in a dog model that underwent a 7-day atrial tachycardia stimulus at a rate of 10 Hz to stimulate AF-related atrial remodeling [
26]. Liu et al. investigated the role of GRP75 (HSPA9) in the modulation of ER-stress in low-dose streptozotocin induced type 2 diabetes (T2DM)-related atrial remodeling in rats. The contact points between the ER and the mitochondria are known as mitochondria-associated ER membranes (MAM). Calcium can be released from the ER via intracellular calcium release channels (IP3Rs) and voltage-dependent anion channels (VDACs) at the outer mitochondrial membrane (OMM), finally moving to the mitochondria. In diabetic atrial remodeling, the IP3R1-GRP75-VDAC1 complex mediates ERS-MAM-mitochondrial oxidative stress and plays an important role in diabetic atrial remodeling. A higher percentage of AF was induced by burst pacing stimulation in the T2DM group than in the control group (75% vs. 14.3%). Silencing or deleting the essential MAM gene GRP75 prevented tunicamycin (ERS inducer)-induced atrial remodeling and AF incidence (18.2% vs. 63.0%) [
25].In addition to ER-stress, oxidative stress is thought to contribute significantly to atrial remodeling and AF promotion. A previous study showed that two membrane subunits of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (p22phox and gp91phox) were significantly activated in the right atrial appendage (RAAs) tissue of AF patients and that these subunits played important roles in atrial reactive oxygen species (ROS) production and AF pathogenesis [
28]. Meanwhile, superoxide dismutase (SOD), the most important antioxidant enzyme family that scavenges ROS, has been reported to be depleted in diabetic atrial tissue, thereby exacerbating the onset of AF [
29]. It has been demonstrated that increasing the expression of mitochondrial HSP70 leads to an increase in ATP production and so regulates mitochondrial activity as well as ROS in cardiomyocytes. HSP70 stimulates mitochondrial SOD and inhibits the nuclear translocation of phosphorylated eukaryotic elongation factor 2 (eEF2) and apoptosis-inducing factor (AIF), resulting in improved mitochondrial function and decreased apoptosis in mice with left anterior descending artery ischemia/reperfusion [
30,
31]. Nobuyuki Murakoshi et.al. investigated the therapeutic benefits of antioxidants and HSP70 on age-related AF. In older rats (9-month-old), AF was induced 17 times out of 18 stimulation procedures (94.4%), compared to 15 times out of 18 stimulation procedures (83.3%) in younger animals (3-month-old). It has been reported that the peroxisome proliferator-activated receptor gamma (PPAR-γ) induces enzymes involved in ROS scavenging. Pioglitazone, a PPAR-γ activator, significantly reduced AF duration in aged rats to 45.6 ± 10.5 seconds (young, 30.7 ± 7.3 second; aged, 107.4 ± 24.2 second) [
32]. Meanwhile, PPAR-γ activator supplementation has been shown to increase HSPA1 mRNA and protein levels in the atria of an AF rat model, inhibit age-related arrhythmogenic atrial remodeling, and AF perpetuation by improving antioxidant capacity and inhibiting the mitochondrial apoptotic signaling pathway [
32]. It should be noted that PPAR-γ activators and HSP70 could be novel upstream therapies for AF.Similar to HSP60, an immediate release of HSP70 into the circulation and a modulation of TLR2 and TLR4 on monocytes after coronary artery bypass surgery (CABG) have been observed [
33]. TLR2 levels were elevated in both monocytes and atrial tissue of patients with AF, and elevated atrial levels of TLR4 have been seen in patients with AF and heart failure [
34,
35,
36]. Increased extracellular HSP70 increased the production of proinflammatory mediators (tumor necrosis factor-α [TNF-α] and interleukin-6 [IL-6]) in a CD14-dependent manner via the TLR2/4-mediated MyD88/NF-κB pathway [
37,
38]. HSP70 treatment of cultured HL-1 cardiomyocytes enhanced the expression of intercellular adhesion molecule 1 (ICAM-1), IL-6, and keratinocyte-derived chemokine (KC) compared to controls [
37]. TLR2 antibodies can prevent contractile dysfunction and cell death by reducing ICAM-1 expression in cardiomyocytes induced by HSP70. Intracellular HSP70s, on the other hand, have been shown to exhibit cardioprotective properties. In myocardial ischemia/reperfusion mice, Dillmann and colleagues found that knocking out HSP70 genes resulted in cardiac hypertrophy after myocardial ischemia/reperfusion injury, which may be related to several signaling pathways, including Jun N-terminal kinase (JNK), p38/mitogen-activated protein kinase (MAPK), Raf-1, and extracellular signal-regulated kinase (ERK) [
39]. The anti-inflammatory impact of HSP70s in AF warrants further research (
Figure 1). 3.3. Small HSPs and Atrial FibrillationCardiomyocytes express large quantities of small HSPs, particularly HSP27, which localize to contractile proteins and the microtubule network, stabilizing the structure and preserving the contractile and electrophysiological properties of cardiomyocytes. Human HSPB1 (Hsp27) levels in RAAs and LAAs tissue samples are reduced in the severe stage of persistent AF [
9]. Overexpression of HSPB1, as well as HSPB6, HSPB7, HSPB8 can independently play a protective role against tachypacing-induced Ca2+ transient reduction via reducing the formation of F-actin stress fibers [
40]. Brundel et al. also demonstrated that phosphorylated HSPB1 (HSP27) could prevent the atrial tachycardia–induced Ca2+ handling, L-type Ca2+ channel current reduction and associated action potential duration (APD) shortening in tachypaced HL-1 cardiomyocytes and isolated canine atrial cardiomyocytes [
41]. Similar protective effects were observed when overexpressing HSP27, which accelerates the recovery of structural damage in AF. HSP27 may shield the contractile proteins from AF-induced cleavage by cysteine proteases (such as calpain 1) [
8,
42]. GGA-59 treatment of HL-1 cardiomyocytes accelerated recovery from the tachypacing-induced restoration of (acetylated) α-tubulin mRNA and protein levels, as well as cardiac troponin I and troponin T [
43]. Several data suggest that HSPB1 protects the microtubule network, possibly through direct binding to histone deacetylases (HDAC) 6. HDAC6 emerges as a key regulator in AF progression by inducing α-tubulin deacetylation and, as a result, calpain-induced microtubule disruption, and may thus be a druggable target in AF [
44]. Tubastatin A and ricolinostat (ACY-1215), two powerful HDAC6 inhibitors, have shown therapeutic effects against microtubule disruption in mouse models of neurological disorders and cancer. Furthermore, tubastatin A protects against electric remodeling (L-type Ca2+ current reduction and APD shortening) and cellular Ca2+ handling/contractile dysfunction, as well as subsequent AF promotion, in a canine model of AF [
45]. These findings suggest that HSPB1 protects the microtubule network, possibly by directly binding to HDAC6 and inhibiting its activity, preventing α-tubulin deacetylation, depolymerization, and subsequent degradation [
43,
46].



留言 (0)