1. IntroductionThe landscape of gene editing has changed in recent years with the advent of Zinc Finger Nuclease (ZFN), Transcription Activator-Like Effector Nuclease (TALEN), and CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR Associated Protein 9) technologies [
1]. Nuclease based gene editing systems hold promise for clinical gene therapy. Several clinical trials have been launched utilizing ZFN (e.g., ClinicalTrials.gov Identifier: NCT00842634), TALEN (e.g., NCT03057912) and Cas9 (e.g., NCT03545815).Cas9 is a promising gene editing nucleases given its ease of use, multiplex capacity, and unparalleled targeting efficiency [
2,
3,
4]. Many groups including us have reported efficient Cas9 mediated gene editing in cultured cells [
5,
6] and animal embryos [
7,
8,
9] for translational biomedical applications [
10,
11]. However, at least three major challenges remain as roadblocks for implementation of clinical gene editing therapeutics. First, off target effects, especially those associated with CRISPR/Cas9 need to be minimized or eliminated [
12]. Second, the homology directed repair (HDR) rate remains low even with the help of gene editing nucleases, limiting the precision of correction based therapeutics [
9]. The third and perhaps most difficult challenge is to realize an efficient and safe in vivo delivery method of gene editing elements, a hurdle for all nucleic acid therapeutics [
13,
14].Issues inherent to current virus-based delivery method include the immune response to the vector itself, oncogenicity due to insertional mutagenesis, the expensive and limited scalability associated with manufacturing, and the relatively small DNA packaging capacity [
15,
16]. There have been resultant cases of cancer and death in ongoing clinical trials [
17,
18], highlighting the safety concerns associated with viral vectors, and prompting an increased interest in the development and utilization of non-viral delivery systems. However, non-viral delivery systems (such as liposomes and polymers) still suffer from low efficacy, although it is generally believed that they have better safety profiles than their viral counterparts [
19,
20,
21]. Although liposome and polymer based gene delivery has made significant progress in in vitro studies and has received deserved attention during the past two decades, these non-viral gene delivery systems are still facing challenges when applied in vivo such as rapid clearance from blood, toxicity, and low efficiency [
22,
23]. A vast improvement in safety and efficiency for in vivo gene delivery systems is required for clinical application [
24,
25,
26]. Polymer nanoparticles such as cationic particles, self-assembling particles, and polymer nanospheres [
27] are an attractive non-viral means for nucleic acid delivery due to their ease of chemical modification, reproducibility, scalability, large capacity for packaging, and increased biosafety [
13,
24,
28]. Previously, we developed a hyperbranched polymer (HP) system for microRNA (miRNA) delivery by attaching polyethylene glycol (PEG) and cationic polyethylenimine (PEI) chains to the outer shell of a hyperbranched polyester molecular core [
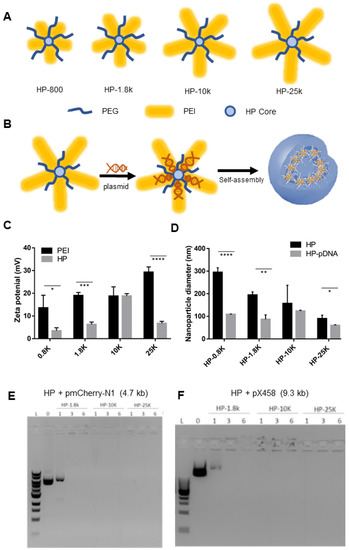
29]. This HP system was demonstrated to be safe and efficient in packaging and delivering miRNA both in vitro and in vivo. Given the engineering flexibility of the HP, we hypothesized that it could be adapted and optimized for enhanced nucleic acid loading capacity to enable packaging and delivering pDNAs encoding gene editing nucleases which are several hundred times larger than miRNAs. Herein, we developed HP polymers bearing different molecular weight polyethyleneimine (PEI) chains (800, 1800, 10K, or 25K Da) and investigated their gene packaging abilities, cytotoxicity, and transfection efficiency. We report that HP-25K nanoparticles can be used to safely and efficiently package and deliver various pDNAs, including those encoding CRISPR/Cas9 and its variant, BE4-max, for gene editing applications. 2. Materials and Methods 2.1. Polymer SynthesisPolyethylenimine (PEI, Mw 800 Da and 25 kDa), polyethylene glycol methyl ether (PEG, Mw 2000 Da), and hyperbranched bis-MPA polyester (64 hydroxyl groups) were purchased from Sigma-Aldrich. PEI (Mw 1800 Da) was purchased from Alfa Aesar (Ward Hill, MA). PEI (Mw 10 kDa) was purchased from Polysciences Inc. (Warrington, PA, USA). All HP polymers (HP4-800, HP4-1.8K, HP4-10K, HP4-25K) were synthesized using methods previously described [
29]. 2.2. HP Solution Preparation
HP was dissolved in DNase and RNase free distilled water (ultrapure water, Invitrogen) in 1–10 mg/mL concentration, and sonicated for 30 min, then centrifuged at 12,000 g for 5 min. The supernatant was used for all HP related experiments.
2.3. Plasmid and Cas9 Construct Preparation
CRISPR plasmids pX330 (#42230) and pX458 (#48138) and EGFP reporter plasmid (#40768) were obtained from Addgene. The mCherry expression plasmid pmCherry-N1 (#632523) was obtained from Clontech. The pDNA was extracted with a “QIAGEN Plasmid Maxi Kit” and the A260/280 ratio was about 1.8.
2.4. Particle Size and Zeta Potential Measurements
The particle size and zeta potential were measured using a Beckman Coulter DelsaNano C Submicron Particle Size Analyzer at room temperature (25 °C). The nanoparticles at various N/P ratios (nitrogen atoms of the polymer to phosphates of DNA) were prepared by slowly adding 60 pmol of DNA (pmcherry-N1 plasmids or pX458 plasmid DNA) solution to an appropriate volume of polymer solution (1.0 mg/mL) and were then incubated at room temperature for 30 min. The HP-pDNA nanoparticles were then diluted with Milli-Q water to a volume of 2.0 mL before measurement.
2.5. Gel Retardation Assay
The HP packaging efficiency was analyzed using a gel retardation assay. Polyplex solution (10 μL) was mixed with 2 μL of 6× loading buffer and loaded on an Ethidium bromide containing 1% agarose gel with Tris-acetate (TAE) running buffer (pH 8.0) and was electrophoresed at 120 V for 20 min. DNA bands were visualized with an ultraviolet (254 nm) illuminator and photographed with a BioSpectrum Imaging System (USA).
2.6. TEM Study on Morphology and Structure of HP-pDNA Nanoparticles
Polyplex solutions were imaged using a negative staining method previously developed in our lab to visualize the pDNA in this work. Tungsten-incorporated pDNA was prepared for use in the nanoparticles. Briefly, 10 mL of sodium tungstate aqueous solution (0.15 mol/L) was added dropwise to 6 mL of HCl aqueous solution (0.8 mol/L) at room temperature. The precipitate was collected and washed to obtain the activated tungstic acid. The desired quantity of activated tungstic acid was added to an aqueous DNA solution with a W/P molar ratio of 3. The W-incorporated pDNA solution was purified by centrifugation and stored at −80 °C. One drop of aqueous polyplex solution (1.0 mg/mL) was added to a carbon-coated copper grid. The grid was dried under ambient conditions. A JEOL 1400Plus TEM was used in this study to obtain TEM images.
2.7. Guide RNAsGuide RNA (gRNA) target sequences were designed using an online tool at Custom Alt-R CRISPR-Cas9 guide RNA tool provided by Integrated DNA Technologies (IDT,
https://www.idtdna.com/ (accessed on 29 November 2022)).
The gRNA sequences are:
TERT sg1: 5′-ACAATCGGCCGCAGCCCGTCAGG-3′;
TERT sg2: 5′-CGCGTACGACACCATCCCCCAGG-3′;
MYLIP sg: 5′-TCTGTACAATGCTGGCGTTGTGG-3′.
PDCD1 sg: 5′-TTAGGGCAGGGCAGGCCGAGGGG-3′.
The PCSK9 gRNA was described in a previous report [
30].
PCSK9 sg: 5′-GGTGCTAGCCTTGCGTTCCGAGG-3′.
The gRNA sequences were cloned in pX330 and pX458 plasmids for transfection experiments as previously described [
31,
32]. 2.8. Cell Viability Assay
Cells: Ad293 cells (Catalog# 240085) were obtained from Agilent. HepG2 cells (Catalog# HB-8065) and Hela cells (Catalog# CCL-2) were obtained from ATCC. Ad293, HepG2 and Hela cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Catalog#11965092, ThermoFisher, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Catalog# SH30071.03HI, Hyclone, Logan, UT, USA) and 1% penicillin-streptomycin (Catalog# 15140122, ThermoFisher). AML-12 cells (Catalog# CRL-2254) were purchased from ATCC, and cultured in DMEM:F12 (Catalog# 11320033, ThermoFisher) supplemented with 10%FBS, 1% insulin-transferrin-selenium (ITS, Catalog# 41400045, ThermoFisher), 40 ng/mL dexamethasone (Catalog# D4902, Sigma-Aldrich, St. Louis, MO, USA), and 1% penicillin-streptomycin(Catalog# 15140122, ThermoFisher). Rabbit fibroblast cells were established in house from ear biopsies collected from New Zealand White Rabbits. The ear biopsy procedure is approved by the University of Michigan Institutional Animal Care and Use Committee (IACUC) protocol #PRO00010094. Rabbit fibroblast cells were cultured in DMEM (Catalog#11965092, ThermoFisher) supplemented with 10% FBS (Catalog# SH30071.03HI, Hyclone), and 1% penicillin-streptomycin (Catalog# 15140122, ThermoFisher).
The cytotoxicity of the polymer was assessed using an MTT assay kit (#CT02, Millipore, Burlington, MA, USA). 1 × 104 rabbit embryonic fibroblast (REF) cells or HepG2 cells were seeded in a 96-well plate one day before encapsulation in the HPs. The amount of HP added to each well was calculated according to 200 ng plasmid DNA multiplied by the N/P ratio. HPs were added to each well accordingly and there were three replicates for each group. The MTT assay was carried out following the manufacturer’s manual.
2.9. Cellular Uptake Assay
Cells (Ad293 (a derivative of HEK293), human (HepG2) or mouse (AML12)) were seeded in a 24-well plate one day before the assay. Upon transfection, 1 µg of pDNA and polymer (HPs or PEIs) at the desired N/P ratio (w/w) were diluted in 50 µL of Opti-MEM (#31985062, Invitrogen, Waltham, MA, USA), incubated at room temperature for 30 min, and subsequently added into each well of cell culture. The control (lipofectamine) transfection process was following the manufacture’s protocol. Briefly, 50 µL lipofectamine dilution in Opti-MEM and 50 µL DNA dilution in Opti-MEM were mixed and incubated for 15 min. Then, the mixture was added into the well of cell culture. The transfection result was obtained after 24 h.
2.10. Flow Cytometry
Cells were washed with PBS and treated with 0.25% trypsin. Cells were then transferred to a 15 mL conical centrifuge tube and spun down at 200 g for 5 min. The pellet cells were re-suspended in 2% FBS in PBS and filtered with a 70 μm Nylon cell strainer (#08-771-2, Falcon, Sydney, Australia). Flow cytometry was conducted at the University of Michigan Flow Cytometry Core.
2.11. Confocal Imaging
Cells were seeded in Chambered Cell Culture Slides (#08-774-25, Falcon), and plasma staining was carried out following the manufacturer’s instructions for CellMask Deep Red Plasma membrane stain (C10046, Thermo Fisher Scientific, Waltham, MA, USA). After removal from the chamber, the slide was mounted in prolong Gold anti-fade mount medium with DAPI (P36931, Thermo Fisher Scientific). Confocal images were taken in the University of Michigan microscopy and image analysis Laboratory with a Nikon-A1 confocal microscope.
2.12. Cas9 Induced Mutation Analysis and QuantificationCas9 induced indel mutations at the target site were analyzed with a T7 endonuclease I (T7E1) assay or by deep sequencing (Deepseq) as we previously described [
6]. The targeted mutation site was amplified via PCR and the PCR products were purified and used for the T7E1 assay. An amplicon of less than 280 bp was amplified before it was sent for deepseq sequencing.
Primers used to amplify target regions for T7E1 are listed below:
TERT sg1
Forward 5′-GCTTCCCCCTAGTCTGTTGTCTGG-3′;
Reverse 5′-CTGGCCCGGCTGCTTCTTGTGGTC-3′.
TERT Sg2
Forward 5′-CCTGACTGCCCGGGCTCCTATT-3′;
Reverse 5′-ACCTCCACCACAGAAACGCATCAC-3′.
PCSK9:
Forward: 5′-CACGGCCTCTAGGTCTCCT-3′;
Reverse: 5′-GCCTCCCATCCCTACACC-3′.
Primers used to amplify target regions for Deepseq are listed below:
TERT sg1
Forward 5′-GGGGCTCAAACGCACTTCT-3′;
Reverse 5′-ACGTCCAGACTCCGCTTCAT-3′.
TERT Sg2
Forward 5′-CGTGAACCTTACGTGGCTCTT-3′;
Reverse 5′-ACTCACACAGGTGGATGTGAC-3′.
MYLIP
Forward 5′-AGGAGAAGCTACGCAAGCTG-3′;
Reverse 5′-AGGAGGGATAGGTGAGGCTG-3′.
PCSK9:
Forward: 5′-CACGGCCTCTAGGTCTCCT-3′;
Reverse: 5′-GCCTCCCATCCCTACACC-3′.
PCSK9 primers amplified amplicons were used for both the T7 endonuclease assay and CRISPR-seq.
2.13. Cas9 Base Editor BE4-Max and gRNA Targeting PDCD1
Cas9 base editor BE4-max plasmid was obtained from Addgene (#112093).
Primers for amplification of targeted region:
Forward 5′-CTTCATCAGGGACTTAGCCTGGC-3′;
Reverse 5′- CAGCCTGGTGCTGCTAGTCTG-3′.
2.14. Statistics
Data are expressed as mean ± standard error of means (SEM) from three replicates in bar graphs, and were analyzed and compared using unpaired, 2-tailed Student’s t test (Graphpad Prism 9.2.0, San Diego, CA, USA). Statistical significance with p < 0.05 is considered significant. Different levels of significant statistical differences were indicated by number of * in each graph, where * p < 0.05, ** p < 0.01, and *** p < 0.001.
4. ConclusionsGene editing nucleases such as Cas9 have become important tools in biomedical research, offering new possibilities to cure or treat monogenetic diseases (e.g., Duchenne muscular dystrophy, Cystic Fibrosis, etc.), and other complex diseases (e.g., AIDS, dyslipidemia, cancer, etc.) [
25]. The clinical application of gene editing therapy requires both efficient and safe delivery of nuclease elements. While more efficient, viral mediated Cas9 pDNA delivery has demonstrated immunogenicity, insertional mutagenesis, and size limitations for DNA cargo [
42].
The present work demonstrates that HP-25K can safely and efficiently transfect multiple cell types with pDNAs of various sizes with low cytotoxicity. Importantly, this system enables Cas9 pDNA delivery to several clinically relevant cell lines, and results in delivery efficiency rivaling and surpassing the commercially available Lipofectamine and PEI25K even for pDNA up to 9.3 kb. HP delivery of pDNA encoding gene editing nucleases can induce indel mutations and efficient BE gene editing in human cells. While further work remains to be done in optimizing the HP nanoparticle system, HPs represent a promising vehicle for the non-viral delivery of CRISPR/Cas9 pDNAs towards the clinical application of gene-editing based therapy.



留言 (0)