
記住我


The TNM classification of cancer is based on the identification of specific histopathologic cancer cell types as well as their origin and invasion into the surrounding tissues, local lymphatic vessels, or distant sites within the human host. Importantly, a number of factors have emerged as markers of poor prognosis, including the presence of a population of highly undifferentiated mitotic figures, an elevated staging score, and an increased level of tumor-specific biological antigens. Nonetheless, a quantitative metric that signifies the inherent virulence of a specific tumor does not currently exist, and the development of such a numeric classifier would complement the qualitative elements that are presently used to categorize individual cancers. To address this, an in vitro model was developed to study the competitive nature of unrelated cancer cell lines, and it was hypothesized that certain super tumors would emerge based on these sequential competitions.
Intercellular competition is a plausible physiological mechanism for optimizing the quality and survival of dissimilar tissue types1. In addition, genes that induce super competition (ie, a level of intercellular competition above that of wild type) have been identified2,3. The capacity to transform normal cells into super competitors is thought to result from the expression of oncogenes of the myc family. In theory, the gene-related transformation of normal cells into a super-competitor state might come at the expense of the surrounding tissue within the tumor microenvironment.
Although the induction of apoptosis in adjacent cells might benefit the predominant tumor cells, the total cell number may remain constant3,4. This phenomenon has been postulated to occur in the early stages of malignant transformation and to potentially account for clinical observations, such as “field cancerization.” In field cancerization, a proliferative advantage is associated with a field of cells of monoclonal origin that expand at the expense of normal tissues and perhaps dissimilar malignant tissues as well5,6. Importantly, intercellular and intracellular communications are generally thought to play central roles in malignant transformation and carcinogenesis, and the biochemical responses of cells to such stimuli in their extracellular microenvironments serve to regulate the intricate biological processes of proliferation, migration, and apoptosis. Notably, the ability to sustain chronic proliferation is one of the most fundamental traits of cancer cells, and under normal conditions, homeostasis of cell number and, thus, maintenance of normal tissue architecture and function are preserved by precise regulation of the production and release of growth-promoting signals that instruct cells to enter and progress through the cell growth and division cycle. Cancer cells become independent of these homeostatic controls via deregulation of extracellular signals, and these signals are typically mediated by the interaction of growth factors with cell-surface receptors, which frequently contain intracellular tyrosine kinase domains. Subsequently, intracellular signals are generated by these tyrosine kinase domains and are propagated via branched intracellular signaling pathways that serve to regulate cell-cycle progression, growth, survival, and energy metabolism. Presently, the nature of the proliferative signals that function within normal tissues remains poorly characterized. In addition, there is little insight into the mechanisms that control cellular proliferation and the release of mitogenic signals7.
The growth factor signaling pathways that regulate cell numbers and their position within tissues are difficult to access experimentally. These extracellular communications are transmitted by way of paracrine signaling in the pericellular and extracellular matrices between cells and are controlled by a complex network of proteases, sulfatases, and other enzymes that liberate and activate signaling molecules in a temporally and spatially regulated manner. The complexity of these networks has limited the precise elucidation of these extracellular mechanisms.
In contrast to our limited insight into such extracellular mechanisms, substantial insight into intracellular mitogenic signaling has been attained8–11. Sustained proliferative signaling in cancer cells may develop in several ways. For example, autocrine proliferative stimulation may result in the formation of cancer cells via the expression of cognate receptors associated with growth factors produces by the cells. In addition, normal cells within the supporting tumor-associated stroma may supply cancer cells with various growth factors in response to signals received from the cancer cells12,13. Sustained proliferative signaling in cancer cells may also result from hyperresponsiveness to available growth factor ligands secondary to increased levels of receptor proteins produced on the surface of the cancer cells. Structural alterations in the receptor molecules that enable ligand-independent activation may also contribute to sustained proliferative signaling. In addition, constitutive activation of signaling pathway components operating downstream of the cell surface receptors may also culminate in growth factor independence, eliminating the need for the restimulation of these pathways by ligand-mediated receptor activation. However, given that multiple downstream signaling pathways may emanate from a single ligand-stimulated receptor, the activation of a specific downstream pathway, such as the pathway that responds to the Ras signal transducer, may only represent a subset of the aggregate regulatory effects of cell-surface receptor activation7.
To further elucidate the mechanisms and cellular processes responsible for mitogenic and proliferative signaling, apoptotic resistance, evasion of growth suppressors, and replicative immortality, the following foundational study was performed, which served to assess the relative dominance of specific cancer cell lines. Moreover, this analysis also lays the foundation for a quantitative metric that would denote the inherent virulence of an individual tumor.
Methods Cell isolationInitially, a number of consecutive surgically excised tumors that had not been previously treated were prospectively harvested, and those specimens determined to be histologically benign, representing a redundant diagnosis, or that had an inadequate sample size were excluded. The remaining samples were all pathologically confirmed to be malignant, and these samples are described in Table 1. Each specimen was visually examined for the location of regions likely to contain the richest concentration of cancer infiltration. Extreme care was taken to avoid areas of necrosis, stromal tissue, thrombotic material, and nests of surrounding normal cells.
Table 1 - Selected tumor samples and demographic characteristics. Symbol Non-Hodgkin lymphoma 73, M L Infiltrative ductal breast cancer 65, F B Gastric adenocarcinoma 53, M G Squamous cell carcinoma of the lung 61, M Lu Renal cell carcinoma 48, F K Ovarian serous cystadenocarcinoma 52, F O Colonic carcinoma 80, M CF indicates female; M, male.
The cancer cells were gently separated from the adherent tissue, placed in a flask, washed with a balanced salt solution, and dissociated with trypsin to produce a cell suspension. A Coulter counter was then used to confirm a >90% rate of dissociation and cell viability. If either could not be confirmed, the process was repeated from the first step until both criteria were satisfied.
Once generated, the cell suspension was diluted with McCoy’s medium 5A to achieve a concentration of 100,000 cells/mL. Next, 10 mL of each tumor cell suspension was subcultured on a bed of prewarmed growth medium and deposited into an incubated stock tumor bank. After a total of seven dissimilar stock tumor subcultures had been collected, ~100,000 cancer cells were withdrawn from each subculture and paired with an equal aliquot of a dissimilar cancer cell line; this process was performed for all possible combinations to yield a total of 21 unique tumor pairs.
Competition assaysThe tumor pairs were plated on diametrically opposed areas along the middle of a gridded petri dish (Fig. 1A). They were then incubated at 37°C for 7 days in a humidified atmosphere of 5% CO2 on enriched growth medium containing 10% fetal calf serum. After which, each dish was then opened and examined, with particular attention paid to movements, migrations, and motility patterns. A relative scoring system was designed that awarded points to those cancer cell lines that exhibited the ability to encroach into the region of its tumor competitor, with additional points accrued for penetration into the more remote areas of the counter-hemisphere (Fig. 1B).
 Figure 1:
Figure 1: A, A gridded petri dish used in this analysis. B, A scoring grid. Points were awarded for passage across the midline and subsequent occupation of a square in the counter-hemisphere.
All of the cellular material on the dish was examined by 3 different board-certified pathologists, blinded to the study design, whose task was to determine the cell type of the material that resided within the square compartments of the dish. Ownership of the compartment was established for a specific cell type if all 3 pathologists were unanimous in their decision. Lack of unanimity resulted in a null allocation for that space.
Within each matched pair, the cancer cell line that accumulated the highest total number of occupation points was declared the winner. If the 2 scores were equal, the match was declared a stalemate. In the case of engagement between gastric and kidney carcinomas, there was no passage of either tumor cell line across the midline; thus, the match was excluded from further consideration.
A table was constructed in descending order of the number of victories (Table 2) to highlight the relative dominance of each cell line. In addition, the margins of victory were calculated to determine the relative strengths, and an alphanumeric modifier was generated using the symbol of the dominant tumor followed by the symbol of the less dominant tumor and then the margin of victory (Table 3). In addition, an aerial-view representation of each battle dish was created to display the final position of each competitor (Table 3; Fig. 2, plates 1–21).
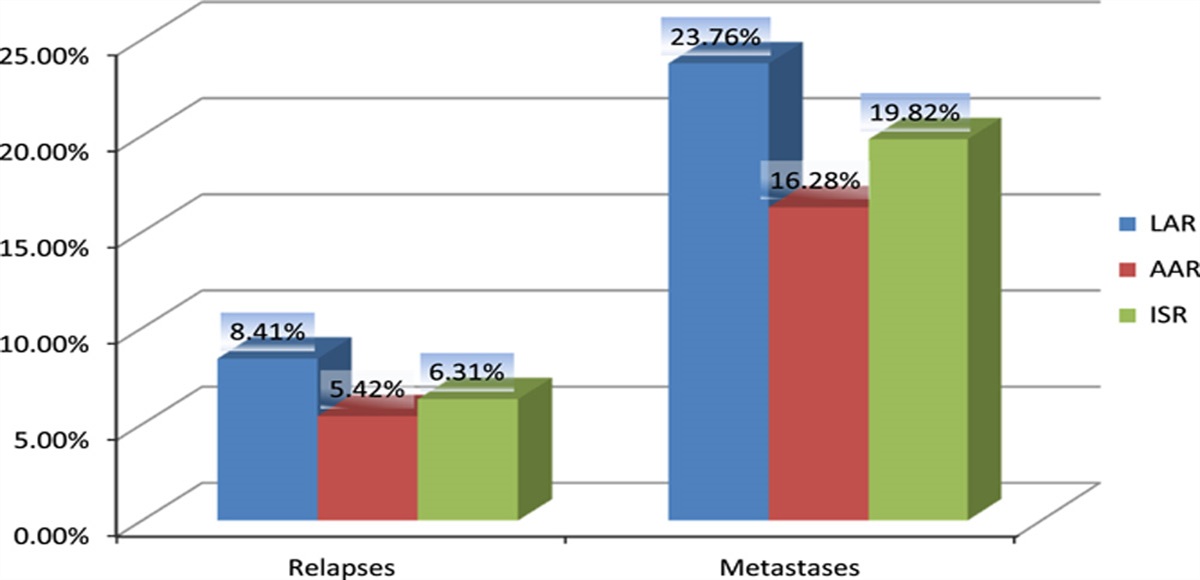
Table 2 - Overall performance of tumor types. Wins Loses Stalemates No Growth Ovarian 5 1 — — Lymphoma 4 1 1 — Lung 4 2 — — Kidney 2 3 — 1 Breast 2 3 1 — Gastric 1 4 — 1 Colon 1 5 — —The margin of dominance between the matched tumors was quantified using an alphanumeric score, and the pairs were rank ordered to convey their relative supremacy.
 Figure 2:
Figure 2: A–D, Aerial-view representations of each battle dish indicating the final position of each competitor. The different cell lines are represented as blue or red, as indicated.
ResultsA total of 20 consecutive surgically excised tumors were prospectively harvested. Among these, 9 specimens were determined to be histologically benign and were excluded from the study. The remaining 11 samples were all pathologically confirmed to be malignant; however, 4 tumors were excluded due to a redundant diagnosis or an inadequate sample size. The remaining 7 samples are described in Table 1.
The tumor pairs were plated onto diametrically opposed areas along the middle of a gridded petri dish (Fig. 1A) and then incubated at 37°C for 7 days. A relative scoring system awarded points to those cell lines that exhibited the ability to encroach into the region of its tumor competitor (Fig. 1B), and a scoreboard was constructed in descending order of the number of victories (Table 2) to reveal the overall dominance of each cell line.
Overall, ovarian cancer, lymphoma, and lung cancer demonstrated general dominance, with wins in at least 2/3 of their matches. Breast and kidney cancer were intermediate in performance, achieving dominance in 1/3 of their matches, and gastric and colon cancer had the poorest performances and were subordinate in the majority of their matches (Table 2).
However, there were several inconsistencies that required reconciliation before a definitive conclusion could be drawn. In the matched-pair competitions (Table 3), the following were observed: lymphoma overwhelmed ovarian cancer, lung cancer dominated lymphoma, and ovarian cancer, although victorious over kidney and colon cancer, achieved these victories by narrow margins. These results indicated the occurrence of relative dominance, as opposed to absolute dominance, with a dominant cell line toward one cell type falling victim to a second cancer cell line of relative lesser dominance, suggesting that there are potentially multiple cellular factors influencing the outcome of each pairing.
DiscussionIn the present study, we demonstrated that intercellular competition resulted in previously untreated ovarian, lymphoma, and lung tumor cells suppressing the growth and proliferation of colon and gastric cancer cells in vitro.
A number of studies have described mechanisms that could theoretically account for intercellular competition and the suppression of tumor cell proliferation and replication. For instance, a scenario in which cellular competition between malignant and nonmalignant cells is mediated by tumor-suppressive miRNAs has been described. miRNAs were found to undergo cellular secretion following exosomal loading14–16. Subsequently, it was found that introduction of antiproliferative miRNAs into the tumor microenvironment by normal cells could impair the proliferation of precancerous cells, whereas cells that have undergone malignant transformation may circumvent this inhibitory mechanism, resulting in carcinogenic growth. Therefore, tumor cells may need to overcome the homeostatic mechanism conferred by tumor-suppressive miRNAs secreted into the tumor microenvironment17.
Similar phenomena may explain the observation that intercellular competition and the emergence of predominant tumor cell types occur in dissimilar subcultured malignant tissues. This occurrence could be due to the same secretory miRNAs or to a variety of secreted chemokines, cytokines, growth factors, or mitogens, as there a numerous cellular functions that could influence diverse tumor cell types when competing for the same pool of resources, including cellular migration, cell-cell contact mediated growth arrest, hyperproliferation, and others.
Importantly, this prospective pilot study was subject to inherent sources of potential bias and a number of limitations. The sample size was small, the observations were limited to gross examination and light microscopy, and the assessments did not include biochemical assays. Although it was not possible to include a control group of nonmalignant cells in the study design, the results demonstrated that these ovarian, lymphoma, and lung cancer cells were the most invasive and aggressive tumor types. These cell types achieved the highest objective degree of infiltration, predominance, and destruction of the opposing tumor cell types within the matched tumor pairs, which had been incubated and positioned onto the square gridded Petri dishes.
Future directionsIt is likely that the future of cancer treatment will evolve beyond its present primitive state, which currently involves the use of broad-spectrum cytotoxic drugs that indiscriminately destroy both normal and malignant cells, toward a more tumor-targeted approach. Moreover, our demonstration that intercellular competition exists between dissimilar cancer cell lines provides substantial insights into the emergence of dominant tumors. It stands to reason that if intercellular competition exists and dominant cell lines can emerge from these competitions, then there must be multiple agents, organic or inorganic, that mediate this process.
The focus of cancer treatment has begun to move towards the identification of the factors that govern both the defensive maneuvers designed to protect the tumor and offensive maneuvers that allow the cancer to repel and/or lyse other cell types. In the future, it is possible that every freshly excised cancer will be cultured and deposited into a target tumor bank. This will allow aliquots of these target tumors to be withdrawn from the target tumor bank and placed in a battle dish to compete against a selection of dissimilar cancer cells that have exhibited repeated, overwhelming dominance against the target tumor type in prior confrontations. The ultimate goal will be the creation of a stock tumor bank that would be loaded with a selection of dominant tumors needed to challenge a freshly obtained target tumor. When one cancer cell line exhibits its dominance over a second dissimilar cancer cell line by a margin of 100 points or greater, then that tumor will be deemed to be a super tumor.
In addition, with the use of proteomics, gas chromatography, and mass spectrometry, we aim to identify the composition of the various protectants, repellants, growth suppressors, and cidal agents that govern the outcomes between the combatants. Once identified and isolated, these agents can be synthetically reproduced for use in research trials to assess their ability to treat cancers in vivo18.
“Survival of the fittest” is the central message of Darwinian theory. On a cellular level, for one tumor to survive when confronted with another, it must be endowed with some advantage. Before this study, we hypothesized that Darwinian theory could be applicable at the cellular level and through sequential competitions certain super-tumors would emerge. Based on the observations presented in this current work, it is clear that some tumors have gained an advantage relative to other cell types; however, whether this advantage is simply inherent to the tissue of origin or, alternatively, conferred by an unknown selective pressure remains to be elucidated.
ConclusionsThis pilot experiment has revealed that intercellular competition exists between previously untreated, subcultured target tumor cells and dissimilar malignant tissues using an in vitro model. Importantly, intercellular competition and the unilateral dominance of select cancer tissue types have not been reported previously in the literature. However, additional studies are needed to validate these findings.
Consent for publicationThis in vitro study utilized cancer specimens obtained from anonymous sources.
Availability of data and materialThe author agrees to submit all available original data to the journal upon request.
Ethical approval and consent to participateThis article does not contain any studies with human participants or animals performed by any of the authors.
Sources of fundingThe study was performed during a fourth-year medical school elective and required no funding.
Author contributionP.W. designed the study, performed the experiments, analyzed the data, and wrote the manuscript.
Conflict of interest disclosureThe author declares that there is no financial conflict of interest with regard to the content of this report.
Research registration unique identifying number (UIN)None.
GuarantorPhilip Weintraub.
References 1. Moreno E, Basler K, Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 2002;416:755–9. 2. De La Cova C, Abril M, Bellosta P, et al. Drosophila myc regulates organ size by inducing cell competition. Cell 2004;117:107–16. 3. Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell 2004;117:117–29. 4. Diaz B, Moreno E. The competitive nature of cells. Exp Cell Res 2005;306:317–22. 5. Donaldson TD, Duronio RJ. Cancer cell biology: Myc wins the competition. Curr Biol 2004;14:R425–7. 6. Secombe J, Pierce SB, Eisenman RN. Myc: a weapon of mass destruction. Cell 2004;117:153–6. 7. Weinberg RA. The Biology of Cancer. Abingdon: Garland Publishing House Science, Taylor & Francis Group; 2006. 8. Perona R. Cell signalling: growth factors and tyrosine kinase receptors. Clin Transl Oncol 2006;8:77–82. 9. Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol 2009;21:177–84. 10. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2010;141:1117–34. 11. Witsch E, Sela M, Yarden Y. Roles for growth factors in cancer progression. Physiology (Bethesda) 2010;25:85–101. 12. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004;432:332–7. 13. Cheng N, Chytil A, Shyr Y, et al. Transforming growth factor-beta signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol Cancer Res 2008;6:1521–33. 14. Gibbons DL, Lin W, Creighton CJ, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev 2009;23:2140–51. 15. Kosaka N, Iguchi H, Yoshioka Y, et al. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem 2010;285:17442–52. 16. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A 2010;107:6328–33. 17. Kosaka N, Iguchi H, Yoshioka Y, et al. Competitive interactions of cancer cells and normal cells via secretory microRNAs. J Biol Chem 2012;287:1397–405. 18. Rubio-Viqueira B, Jimeno A, Cusatis G, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res 2006;12:4652–61.
留言 (0)