
記住我
Inflammation is a complicated case of physiological changes caused by external causes that presents considerable hurdles in terms of treatment. Due to the rise of drug resistance and negative responses to synthetic anti-inflammatory medications, efforts have commenced to explore herbal remedies known for their rich resources, minimal side effects, and positive treatment outcomes. The advancement of natural anti-inflammatory treatments has become a point of research interest [9, 18,19,20].
COX-1, 2, and 5-LOX inhibition assaysAll this was already mentioned earlier-delete Evaluation of the potential COX inhibitory effect of the crude extract of the crude leaf extract of D. erecta and its fractions showed that the crude extract and the methylene chloride and ethyl acetate fractions had a COX inhibitory effect, with a strong preference for COX-2 (Table 1). Bioactivity-guided isolation yielded Duranterectoside A (1) from both bioactive fractions, suggesting that it may be the primary component responsible for the observed bioactivity in the plant. Not enough evidence to support this; it might just be a matter of polarity of the metabolite.
Table 1 IC50 values of methanolic extract and its respective fractions of D. Erecta Linn on COX-1, 2, and 5-LOX enzymes lettering of extract and fractions DO NOT coincide with those in the experimental sectionSpectral data of the isolated compoundsThe phytochemical analysis of the TE using various chromatographic techniques resulted in the separation of three compounds (compounds 1–3) (Fig. 1 and Table S1). The isolated compounds were identified as two iridoids glycosides and one flavone aglycone that were identified based on their structures using spectroscopic data (ESI-MS, 1H-NMR, and 13C-NMR), as well as comparisons to published data (Figs. S2–S10).
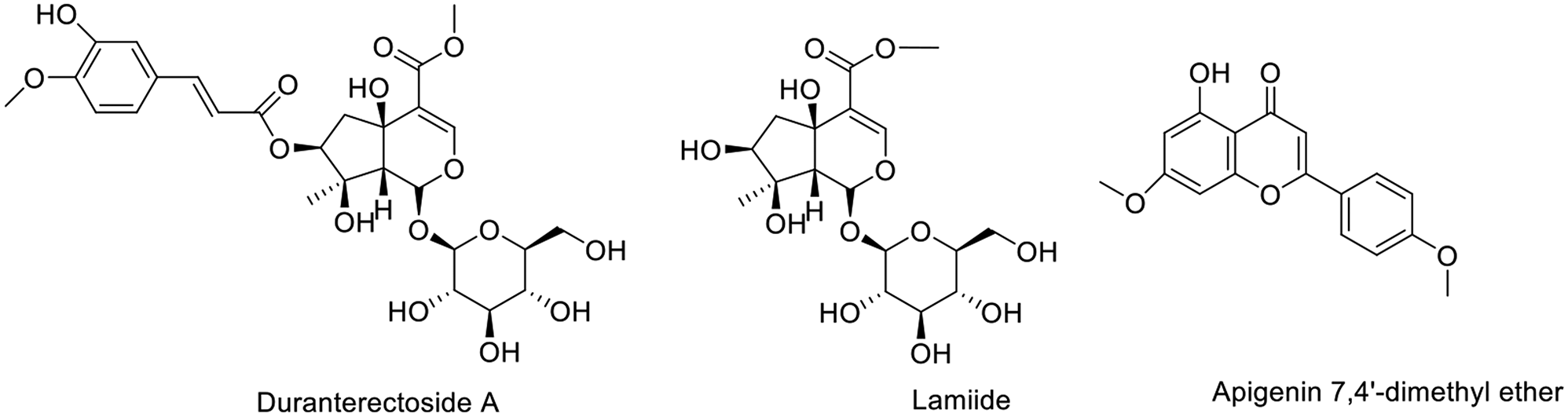
Fig. 1Structures of isolated compounds. Numbers should be included, in parenthesis, next to the names
Compound 2 was isolated as white powder. The molecular formula of compound 2 was determined by negative ion ESI-MS: m/z 421.2350 [M − H]− for C17H25O12 and 467.24320 ([M + HCOO]− for C18H27O14, in agreement with 17 observed resonances in the 13C-NMR spectrum (Table S1 and Figs. S2–S4). The 1H-NMR spectrum of 2 (Table S1, Fig. S3) displayed δH 7.46 (1H, s, H-3); 3.758 (s, MeOOC), a CH2 group (δH 2.32 (1H, dd, J = 16.1 and 1.7 Hz, H-6a) and 2.35 (1H, dd, J = 16.1 and 5.0 Hz, H-6b); an oxymethine (δH 4.77 (1H, dd, J = 5.0 and 1.7 Hz, H-7); and a Me group (δH 1.06 (3 H, s, H-10), a characteristic signals for a C-10 iridoid. The anomeric sugar proton at δH 4.56 (1H, d, J = 7.9 Hz, H-1‵), indicated the presence of a β-glucose unit. Both the chemical shifts and coupling constants of H2-6 and H-7 of 2 suggested that the 7-OH group was β-oriented. From these data (Table S1), the structure of compound 2 was identified as lamiide [21, 22].
Compound 1 was isolated as white powder, and determined through negative ESI-MS (m/z 597.3323 [M − H]− for C27H33O15). The 1H and 13CNMR spectra of 1 were closely resembled to those of lamiide (2), with the exception of a downfield shift (ca. 1.49 ppm) for H-7, which appeared at 4.77 in 1, along with a downfield shift (ca. 3.4 ppm) for C-7, together with the upfield shifts for C-6 and C-8, indicating the presence of an acylated oxygen at C-7 in 1. Moreover, signals indicative of a trans-isoferuloyl group were detected at C-7. Hence, it was confirmed that duranterectoside A (1) is the 7-O-trans-isoferuloyl ester of lamiide [23].
Compound 3 was isolated as yellow powder, and the molecular formula was determined through negative ion ESI-MS, showing m/z 297.2365 [M − H]− for C17H13O5. In the 1H NMR spectrum of 3, two singlets were observed at δ 3.63 (3H, s) and 3.75 (3H, s), attributed to the two methoxy groups at positions 4’ and 7’, respectively. Additionally, signals were detected at δ 6.42 (1H, s, H-3), two proton doublets at δ 7.69 (2 H, dd, J = 7.7, 2.2 Hz) and 7.09 (2 H, dd, J = 7.7, 2.2 Hz), corresponding to H2’/H6’ and H3’/H5’ protons. Two doublets at δ 6.28 (1H, brs) and 6.35 (1H, brs) were assigned to protons H-6 and H-8, respectively. In the 13C NMR spectrum of 3, 15 signals were observed for 17 carbons. A signal at δ 182.37 was designated for C-4, while signals at δ 56.93 and 59.63 were linked to two methoxy groups at C-4 and C-7. Furthermore, signals at δ 127.95 and 115.65 were assigned to carbons C-2/6 and C-3/5, respectively. Signals at δ 101.16 and 94.18 were attributed to C-6 and C-8. Based on the spectral data provided, compound 3 was identified as apigenin 7,4’-dimethyl ether [24].
In-vitro anti-inflammatory assaysTwo iridoids, duranterectoside A (1), lamiide (2), and apigenin 7,4’-dimethyl ether (3), were tested against the three enzymes (Table 2). Iridoids are the primary bioactive principles in Duranta spp., with potent pharmacological and biological activities such as anti-inflammatory, antioxidant, neuroprotective, cardioprotective, antitumor, hypoglycemic, hypolipidemic, antiallergic, antimalarial, antibacterial, antiviral, and insect repellent properties [25]. Lamiide (2) has been previously reported to have anti-inflammatory properties in carrageenan-induced rat paw edema and rat brain phospholipid tests, suggesting that 2 may exert its anti-inflammatory action by scavenging free radicals from the lipid membrane [26]. Similarly, it has been reported that lamiide (2) suppresses soybean 5-LOX, when tested in vitro [21] and that possesses a moderate inhibition of lipoxygenase at a dose of 0.5 mM [27]. Collectively, iridoids are reported to have anti-inflammatory activity by inhibition of phospholipid peroxidation, leukocyte accumulation and influx, free radical scavenging activity, and their inhibitory power against 5-lipooxgenase enzyme, in addition to histamine and bradykinin release [22]. Apigenin 4’,7-dimethyl ether, a methylated flavone, has been shown to have strong anti-inflammatory properties mainly due to its prostaglandin inhibitory potential [7]. Our findings indicate that the three isolated compounds exhibit anti-inflammatory activity, primarily mediated by their selective inhibition of COX-2, along with potential inhibitory effects on 5-LOX.
Table 2 IC50 values of the isolated compounds (1–3) on COX-1, -2, and 5-LOX enzymesIn silico studiesDiscovery Studio 4.0 software was used for in silico molecular docking, dynamic modelling, and ADMET experiments.
Molecular dockingMolecular docking studies are a powerful method for understanding the various interactions between ligands and the active sites of enzyme. The in-vitro anti-inflammatory activity of isolated compounds 1, 2, and 3 against COX-1, 2, and LOX prompted us to conduct a molecular docking study to interpret the biological results and gain a better understanding of the binding poses and interactions with the key amino acids in each enzyme’s binding site. Celecoxib was co-crystallized with COX-1 and COX-2, which were obtained from the protein data bank (PDB ID: 3KK6) [14] and (PDB ID: 1CX2) [15] and gallocatechin co-crystallized with the LOX (PDB ID: 1JNQ) [16] were used as reference compounds to evaluate the molecular modeling docking study results. To validate the C-Docker methodology used in this investigation, the lead chemical was re-docked into the active sites of each enzyme. The results showed good agreement, with RMSD values of 0.5 Å, 0.4 Å, and 6 Å for COX-1, COX-2, and 5-LOX, respectively. These results support the efficacy of the docking approach used [28]. The presented docking study revealed only comparable binding mechanisms between the lead drug and the docked molecules, particularly for COX-2. Table 3 summarizes the CDOKER-interaction energy and the drugs’ interactions with critical amino acids via COX-2. The molecular docking of the three compounds using the C-Docker methodology revealed that compounds 1 kept two essential H-bonds with TYR355 and ARG120 in COX-2 and one essential H-bond with GLN192 in COX-1 active site compared to the reference compound celecoxib (Fig. 2). It is worth noting that the phenylacryloyl moiety of compound 1 enhanced its interaction with COX-2 and LOX, where it revealed a hydrogen bond with Arg513 and carbonyl oxygen of COX-2. In addition, the substituted phenyl ring lay in a deep extended hydrophobic pocket through hydrophobic interaction with Asp515 and Gly354. Furthermore, Compound 1 demonstrated greater docking scores than the docked lead compounds. These findings helped to explain why compound 1 had such a strong inhibitory effect when compared to the other derivatives.
Table 3 The C-Docker interaction energy and key amino acids interaction for COX-2Fig. 2(A, C) Retrieved docking pose of Celecoxib with COX-2 and COX-1, respectively showing the key interactions as reported. (B, D) Docking poses of compound 1 with COX-2 and COX-1, respectively. (E, F) docking pose of compound 2 with COX-2 and LOX, respectively, (G) docking pose of Zileuton with LOX
Standard dynamic simulationMolecular dynamic simulations were carried out to investigate the ligand-protein interaction in motion that contributes to their stable bound conformation and to visualize the influence of ligand binding on protein conformational changes [10]. To better understand the action of compound 1 on COX-2, MD simulations were done first on the free protein COX-2 (without any ligand), followed by MD simulations of COX-2 in contact with compound 1.
The RMSD (Fig. 3c), root mean square fluctuations (RMSF) (Fig. 3e), and energy (Fig. 3a) were investigated for free Protein COX-2, and their contributions were plotted as a time-dependent function of MD simulations. The RMSD of the protein backbone was found to fluctuate around 4Å (Fig. 3c). These averaged constant graphs indicate that the protein structure remains steady during each 200 ns simulated time period. The RMSF of all residues was determined during the 200 ns MD simulations to identify the protein’s higher flexibility areas. In COX-2 RMSF graph (Fig. 3e), significant peaks of fluctuations have been seen with the initial 50 residues with over 9.8 Å, residue number 120 up to 5.6 Å, residue 360 with 4.5 Å, and between 400 and 550 with up to 3.0 Å. Finally, we have analyzed the energy involved for the stabilized conformation of this protein and it was observed to have an averaged − 17,250 and − 17,600 kcal/mol of energy (Fig. 3a). Taking all of the foregoing observations in RMSD, RMSF, and energy contributions into account, it is possible to conclude that the COX-2 protein structure remains rather stable throughout the simulation. MD simulations of COX-2 in complex with compound 1 were carried out to better understand the effect of compound 1 binding to COX-2. MD simulations were conducted using the best docked pose of the COX-2-compound 1 binding complex with a binding energy of − 55.5 kcal/mol obtained from protein-ligand docking (Fig. 2). The results are given on Fig. 3. Figure 4d shows that the RMSD of the trajectories for COX-2 in complex with 1 fluctuates around 3.0 Å.
These findings strongly indicate compound 1’s substantial inhibitory and stabilizing capabilities on COX-2 when compared to COX-2 residue variations in the absence of ligand, which were found to exhibit highly fluctuating peaks as described in the preceding section. The 200 ns simulation time used in this investigation is sufficient to allow for side chain rearrangements in both the natural and protein-compound 1 complexes, resulting in the most stable binding configuration [15]. In addition, RMSF was determined for each residue index to assess the flexibility of the COX-2-compound 1 complex, which exhibited a significant decrease in fluctuations compared to the free protein, indicating improved stability (Fig. 3f). After analyzing the total energy involved for the stabilized conformation of this protein in complex with 1, we found that it maintains an average of − 17,200 and − 17,550 kcal/mol of energy (Fig. 3b), which is well minimized in contrast to COX-2 in the absence of ligand.
Fig. 3(A) Total energy Vs time in production step of free protein, (B) Total energy Vs time in production step during interaction of compound 1 best conformation poses with protein COX-2. (C) RMSD for free protein (D) RMSD for compound 1-COX-2 complex. (E) RMSF for free protein. (F) RMSF for compound 1-COX-2 complex
In silico predictive ADMET studyThe ADMET study focuses on the chemical structure of the molecule and includes the calculation of several parameters using Discovery Studio 4.0 software: blood-brain barrier level, absorption level, atom-based log P98 (A log P98), 2D polar surface area (ADMET 2D PSA), Cytochrome P450 2D6 (CYP2D6), hepatotoxicity probability, aqueous solubility level, and plasma protein binding logarithmic level (PPB Level) (Fig. 4; Table 4). Compounds 1 and 2 in the ADMET plot had BBB levels of 4, indicating that they cannot breach the blood-brain barrier and so will not induce CNS adverse effects. While compound 3 can pass through the BBB. Compounds 1 and 2 are expected to have limited intestinal absorption, but compound 3 demonstrated high absorption (Fig. 4). All of the substances had an ADME aqueous solubility level between 3 and 4, indicating acceptable solubility. The CYP2D6 score indicates whether a specific chemical structure inhibits or does not inhibit the Cytochrome P450 2D6 enzyme. Compounds 1 and 2 are predicted to be non-inhibitors of CYP2D6, hence side effects such as liver dysfunction are not expected with their administration, as opposed to compound 3. The plasma protein-binding parameter indicates the degree to which a drug binds to carrier proteins. These chemicals can be anticipated to reach the desired targets because they all showed 90% plasma protein binding. The main property (PSA) is a determinant influencing medication bioavailability. Thus, passively absorbed compounds with PSA levels greater than 140 are assumed to have low bioavailability. Compounds 1 and 2 have PSA values greater than 140, indicating limited passive oral absorption, with the exception of compound 3, which has a PSA value of 64.906 and good bioavailability.
Table 4 ADMET predictions of the compounds 1, 2 and 3Fig. 4The ADMET plot uses calculated PSA_2D and A log P98 properties
留言 (0)