
記住我
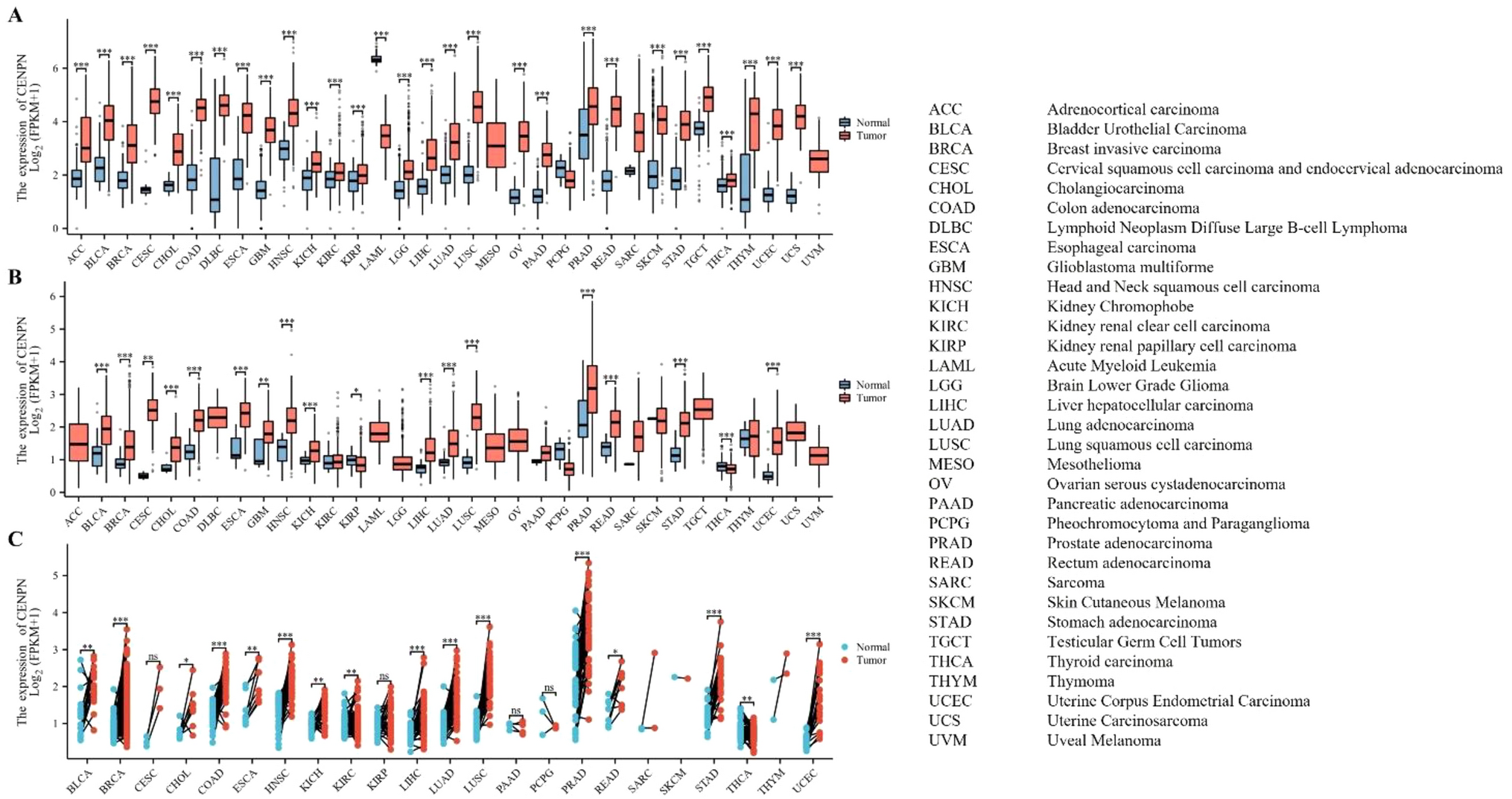



Through the ssGSEA analysis, we figured out the Tregs activity score of KIRC. Based on the optimal cut-off value, the KIRC samples were separated into high- and low-Treg activity groups. We observed a poor prognosis that occurs in high Tregs activity groups (Fig. 1A), which lures us to further explore the potential reason and mechanism of Tregs on KIRC prognosis. The first thing we did was to determine the Tregs-related genes. To do this, we calculated the Pearson correlation coefficients (PCC) between each gene expression and Tregs score based on the whole genome expression profile of KIRC(TCGA). Those with absolute PCC > 0.4 and P-value < 0.05 were identified as Treg-related genes (Fig. 1B). Then we carried out differential expression analysis by comparing the tumour with the normal expression profile of KIRC with a filtering threshold based on absolute log2(Fold Change) > 2 and adjusted P-value < 0.05, differential expression genes (DEGs) were finally screened (Fig. 1B). 76 KIRC Tregs-related genes (KTRGs) were eventually determined by overlapping Treg-related genes and DEGs. The heatmap revealed that these genes' expressions could rightly discriminate between tumour and normal samples (Fig. 1C). When the KTRGs expression profile was conducted dimensionality reduction by t-distributed Stochastic Neighbor Embedding (t-SNE), we also observed tumour and normal samples harboured in different areas in a two-dimension axis, indicating that the expression of these genes could distinguish the tumour and the normal tissues well (Fig. 1D). We then presented the mRNA expression profile of KTRGs to clinical survival data, and a univariate Cox regression analysis was subsequently performed. 22 genes of 76 KTRGs were estimated to be associated with the overall survival of KIRC. Among these, SPI1, TYMP, LILRB1, IL20RB, ITGAX, C1QL1, FCER1G, SLAMF8, TGFBI, CCL5, and CXCL13 were risky genes to the prognosis of KIRC patients, high expression of which represent a worse survival probability (Fig. 1E and Figure S1). While OGDHL, MTURN, HSD11B2, PPARGC1A, LDHD, FREM1, NE3C2, TRIM2, SLAMF8, AGPAT9, and WDR72 were protective genes to OS of the patients and low expression of which represent a worse clinical outcome. In the protein–protein interaction (PPI) network, HSD11B2, NR3C2, and PPARGC1A show a protein interaction, SLAMF8, FCER1G, ITGAX, LILRB1, SPI1, CCL5, and CXCL13 show a protein interaction, while others do not show a relationship (Figure S2A). In mRNA levels, WDR72, MTURN, NR3C2, AGPAT9, TRIM2, PPARGC1A, OGFHL, LDHD, FREM1, and HSD11B2 show positive correlation with each other and negative correlation with other genes (Figure S2B). We further analyzed the epigenetic characterization, and we found that the mutation rate of all KTRGs is low (i.e., mutation rate < 5%), but the copy number variation (CNV) of some is exuberant (Figure S2C and D). Among these, TGFBI, SLAMF8, MTURN, LILRB1, LDHD, ITGAX, IL20RB, and HSD11B2 show high amplification (Amp) rate, TRIM2, OGDHL, NR3C2, IL20RB, FCER1G, and ALDH6A1 show high deletion (Del) rate. The expression of IL20RB, TRIM2, NR3C2, CXCL13, SLAMF8, and FCER1G are affected by copy number variation, while other genes do not show a significant effect.
Fig. 1
Identification of KIRC Tregs-related genes. A Kaplain-Meier survival curve reveals the survival difference of the patients in high and low Tregs activity groups. B Schematic shows the process about screening KTRGs. C Heatmap shows the expression difference of KTRGs between tumor and normal groups. D The t-SNE analysis based on the expression of KTRGs between normal and tumor tissues. E The forest plot shows the prognosis-related KTRGs determined by the univariate Cox regression analysis
3.2 Clinical relevance and mechanisms of KTRGsApart from the relationship between prognosis and KTRGs mentioned, we matched the clinical stage and grade data and mRNA expression profile of KTRGs. Analysis of variance (ANOVA) was performed to estimate the difference of genes in different groups (i.e., Grade I—IV). Gene expression levels were shown by heatmap. We found OGDHL, PPARGC1A, LDHD, AGPAT9, ALDH6A1, WDR72, HSD11B2, FREM1, MTURN, NR3C2, and TRIM2 decrease little by little with the advance of grade and stage, whereas TGFBI, IL20RB, C1QL1, CCL5, CXCL13, TYMP, ITGAX, LILRB1, SLAMF8, SPI1, and FCER1G, gradually increase with development of disease (Fig. 2A and B). Through analyzing the correlation between 50 hallmark pathway activities and KTRGs expression, we found several genes (i.e., TGFBI, SPI1, SLAMF8, LILRB1, etc.) expression positively correlated with cancer-related pathways such as G2M checkpoint, E2F targets, Hedgehog signalling, Wnt pathways, P53 pathways, etc. PPARGC1A, OGDHL, ALDH6A1, and AGPAT9 have a positive correlation with adipogenesis, fatty acid metabolism, heme metabolism, bile acid metabolism, androgen response (Fig. 2C). PROGENy algorithm (Pathway RespOnsive GENes) concludes several cell-specific signalling pathways [15]. Consistently, TGFBI, SPI1, SLAMF8, LILRB1, FCER1G, CXCL13, CCL5, and C1QL1 show a positive correlation with most cancer-related pathways. WDR72, TRIM2, PPARGC1A, FREM1, ALDH6A1, and AGPAT9 show a negative correlation with most cancer pathways but a positive correlation with androgen pathways (Fig. 2D). All in all, these KTRG expressions are related to tumour progression and might function differently by regulating specific metabolisms.
Fig. 2
Clinical relevance and mechanism analysis of KTRGs. A The heatmap shows the expression of KTRGs across different grades. B The heatmap shows the expression of KTRGs across different stages. C The heat map shows the correlation between KTRGs and activity scores in cancer-related hallmark pathways. D The heat map shows the correlation between KTRGs and the activity score of the progery pathway signature. The red bubble represents a positive correlation; the blue bubble represents a negative correlation. The size of the bubble shows significance; the larger the size, the more significance it has. *P-value < 0.05; **P-value < 0.01; ***P-value < 0.001; ****P-value < 0.0001
3.3 KTRGs risk signature constructionTo further identify the potential important KTRGs, we matched the KTRGs expression profile with KIRC clinical data, and then the data was submitted to the Boruta algorithm. Three types of KTRGs, including confirmed (i.e., important) genes, tentative genes (i.e., probably important), and rejected (i.e., unimportant) genes, were finally determined. Among these, CXCL13, WDR72, TRIM2, ALDH6A1, NR3C2, PPARGC1A, and IL20RB were defined as confirmed genes, SLAMF8, FCER1G, LDHD, MTURN, TYMP, and OGDHL were defined as tentative genes, while others were considered as rejected genes (Fig. 3A). Then, we separated the KIRC(TCGA) datasets into two cohorts, training and testing cohorts, according to the ratio of 7:3. We dismissed the rejected genes and abstracted the expression profile of confirmed and tentative genes from the training cohort to conduct lasso-cox regression analysis. Five genes were finally screened, including NR3C2, WDR72, ALDH6A1, CXCL13, and IL20RB. Interestingly, the genes identified by the lasso-cox algorithm are all confirmed genes, indicating the importance of these genes for prognosis under these two algorithms. Based on the lasso coefficient per gene, a risk score was finally calculated according to the equation:
$$} = \, 0.0 \,*} - \, 0.\,*} - \, 0.00\,*} - \, 0.0\,*} + \, 0.0\,*}0}$$
Fig. 3
KTRGs risk model construction. A Schematic shows the process about constructin KTRGs risk model. B and C Kaplain-Meier survival curve shows the overall survival distinction between high and low KTRGs risk model in KIRC(TCGA) training and testing cohort. Time-dependent ROC curve shows predictive accuracy of risk model in 3-/5-/7-year prognosis. Heatmap shows the KTRGs risk signature expression in high and low KTRGs risk groups
According to the ideal cut-off value estimated by the surv_cutpoint function from the survminer R-package, the KIRC training cohort was divided into high and low-expression groups. Survival curve analysis shows that high risk group patients are faced with the problem of low survival probability (Fig. 3B). From ROC curve analysis, we perceived the area under curves (AUC) at 3-/5-/7-year were 0.6915, 0.7025, and 0.7050, indicating that the KTRG risk score is highly accurate in predicting the prognosis of KIRC patients. The heat map shows that NR3C2, WDR72, and ALDH6A are highly enriched in the low-risk group, while CXCL13 and IL20RB are highly enriched in the high-risk group. The same analysis was performed in KIRC testing cohort, and we observed a similar result (i.e., For ROC, AUC at 3 years = 0.7212, AUC at five years = 0.7392, AUC at 7 years = 0.7107) (Fig. 3C). We utilized other two RCC datasets to further validation, and the results bear resemblance to KIRC datasets (i.e., For E-MTAB-1980 ROC, AUC at three years = 0.7309, AUC at five years = 0.7455, AUC at 7 years = 0.6919; For KIRP ROC, AUC at three years = 0.5940, AUC at five years = 0.6196, AUC at seven years = 0.6097) (Figure S3A and B). These results revealed that the KTRG risk score is practical and accurate for predicting outcomes of patients with kidney cancer.
3.4 Clinical relevance and robustness of KTRGs risk scoreTo further explore the relationship between KTRG risk score and clinical variables, we matched the clinical variables (i.e., pathological stage, histological grade, Gender, and Age) with a 5-gene KTRG expression profile in two RCC cohorts. We found that KTRG risk groups were not correlated with patients' age in the KIRC cohort but correlated with stage and grade, appearing that more advanced stage and grade harboured in high-risk groups (Fig. 4A). CXCL13 and IL20RB are highly enriched in high-risk groups, indicating that the expression of these two genes might promote disease progression. We then divided the clinical data into different clinical sub-groups and matched them with the corresponding mRNA expression profile of KTRGs. Intriguingly, we found that, whatever the subgroup is, patients' outcomes in the high-risk group are always poorer, indicating that the KTRG risk score is robust and steady, which could predict the prognosis of patients in every sub-classes of kidney cancer (Figure S4). The same analysis was performed in the E-MTAB-1980 cohort, and we observed the same results (Fig. 4B and Figure S5).
Fig. 4
Clinical relevance of risk model. A Heatmap shows the five gene expressions in high and low KTRG risk groups and the distribution of clinical variables (including pathological stage, histological grade, gender, age, OS status, and OS time) in high and low KTRGs risk group in KIRC(TCGA) dataset. B Heatmap shows the 5 gene expression in high and low KTRG risk groups and the distribution of clinical variables (including pathological stage, histological grade, gender, age, OS status, and OS time) in high and low KTRGs risk group in the E-MTAB-1980 dataset. *P-value < 0.05; **P-value < 0.01; ***P-value < 0.001; ****P-value < 0.0001
3.5 Clinical nomogram model constructionTo develop the clinical nomogram, we initially analyzed the correlation between various clinical variables and the prognosis of patients with KIRC (Figure S7A). Our analysis revealed that additional pharmaceutical therapy, additional radiation therapy, age, history of neoadjuvant treatment, tumor size, grade, stage, chemotherapy outcomes (Progressive Disease [PD], Stable Disease [SD], Complete Response [CR], Partial Response [PR]), laterality, and risk score are significantly associated with patient prognosis. However, we observed that patients who received additional pharmaceutical therapy, radiation therapy, and neoadjuvant treatment exhibited a higher risk for poor outcomes, which contradicted our expectations. Upon further investigation into missing clinical data, we identified several variables with a high rate of data absence (> 20%) (Figure S7B). To ensure data integrity and reflect clinical reality, we selected a refined set of variables for further analysis, including age, risk score, gender, grade, laterality, and stage. In a multivariate Cox regression analysis, we identified the Kidney Cancer Risk Group (KTRG) risk score, grade, age, and stage as independent prognostic factors (Figure S7C). The E-MTAB-1980 dataset, which only included age, gender, grade, and stage variables, was used to perform a multivariate Cox regression analysis, reaffirming that the KTRG risk score remained an independent prognostic factor (Figure S7D). Subsequently, we subjected the variables (age, stage, grade, and KTRG risk score) from the KIRC dataset to stepwise regression analysis. Based on the lowest Akaike information criterion (AIC) value within the KIRC (TCGA) dataset, these four variables were deemed suitable for constructing a clinical nomogram model to predict the 3-, 5-, and 7-year survival probabilities (Fig. 5A). The calibration curve and ROC curve revealed that the model possesses a high discriminative accuracy (AUC at three years = 0.8081, AUC at five years = 0.761, AUC at seven years = 0.7394) (Fig. 5B and C). The nomogram risk score was further calculated based on the established model, and the high and low model risk scores showed extremely significant differences in overall survival probability (i.e., after 100 months, the survival probability of patients with a high model risk score is proximately zero, while in low model risk score are fifty per cent or so) (Fig. 5D). The result reveals that the nomogram risk model could stratify the patients with different prognoses more effectively and discriminatively. This is indeed the case when observing decision curve analysis (DCA). The nomogram model shows a maximum benefit in predicting a 3-/5-/7-year prognosis of patients compared to other single variables (Fig. 5E). The same analysis recurred in E-MTAB-1980 datasets, and the results are consistent with KIRC(TCGA) dataset (i.e., for ROC curve, AUC at three years = 0.8974, AUC at five years = 0.8805, AUC at seven years = 0.8241; for survival curve, the survival rate of patients in high-risk group is forty per cent while in low-risk group are eighty per cent about, after fifty mouths) (Figure S6).
Fig. 5
Nomogram model construction and model evaluation in the KIRC(TCGA) dataset. A The nomogram model (including risk score, age, stage, and grade) predicts 3-/5-/7-year survival probability in the KIRC(TCGA) dataset. B and C Calibration and time-dependent ROC curve of nomogram model. D The survival curve shows the prognostic difference between the high and low-risk groups based on the nomogram model. E Decision curve analysis compares the predictive clinical benefit of risk score, age, gender, grade, stage, and nomogram model for a 3-/5-/7-year prognosis
3.6 Differentially enriched pathways between high and low KTRGs risk groupsGiven the differential outcomes of the high and low KTRG risk groups, we try to explore the potential distinct mechanisms between these two groups. We first submitted the mRNA expression profile of KIRC(TCGA) to gene set enrichment analysis (GSEA) software, and then the potential cancer-related hallmark pathways were further explored. We found several pathways highly enriched in the high KTRGs risk group, and the top three are the EMT pathway, Allograft rejection, and IL6 JAK STAT3 signalling (Fig. 6A). Several metabolism-related pathways are highly enriched in the low KTRGs risk group, such as heme metabolism, fatty acid metabolism, and adipogenesis (Fig. 6B). Further analysis combining the pathways activity score and risk score to conduct Pearson correlation coefficient, the results represent that KTRGs risk score highly positively correlated with MYC TARGET V2, IL6 JAK STAT3, ALLOGRAFT REJECTION, DNA REPAIR, and UNFOLDED PROTEIN RESPONSE, and negatively correlated with FATTA_ACID_METABOLISM, PROTEIN_SECRETION, ADIPOGENESIS, ANDROGEN_RESPONSE, HEME METOBOLISM, etc. (Figure S8). When analyzing the biological process, we found several immune processes highly enriched in high KTRG risk groups, including antimicrobial humoral response, antibacterial humoral response, and regulation of acute inflammatory response to antigenic stimulus (Fig. 6C). Furthermore, the metabolism process, in terms of lipid modification, lipid oxidation, and negative regulation of the amide metabolic process, is mainly enriched in low KTRG risk groups (Fig. 6D). Considering the metabolism-related pathways and processes consistently differential enrichment, we collected some metabolism-related signatures such as energy, amino acid, vitamin, nucleotide, carbohydrates, and TCA and submitted them onto GSEA software. The results are consistent, and all these metabolism signatures are highly enriched in low KTRG risk groups (Fig. 6E and F). These results suggested several hallmark pathways, metabolism signatures, and some immune mechanisms occurred differentially enrichment between the two risk groups.
Fig. 6
Pathways enrichment analysis. A-D Top 6 enriched cancer-related hallmark pathways and biological process in high KTRGs group. E and F Metabolism-related pathways enrichments between high and low KTRGs groups
3.7 Immune relevance and immune therapeutic response of risk scorePrevious research pointed to the effect of Tregs on the immune system and even on immune therapeutic response. Respecting the differential enrichment of the immune process mentioned above we had analyzed, we further explored the relevance of immune and KTRG risk scores. We found the fraction of some anti-tumor immune cells, such as T cells CD8 and T cells CD4, is higher, while B cell naive, NK cell activated, and M1 macrophages are lower in the high KTRGs risk group. Needless to say, the Tregs fraction is higher in the high-risk group. However, the pro-tumor immune cells—M2 macrophage fraction decreased in the high-risk group (Fig. 7A). For immune cell activity, interestingly, B cells, CD8 + T cells, NK cells, Cytotoxic cells, and Tregs consistently increase in high-risk groups. There is no doubt that the KTRG risk score shows the highest positive correlation with the Tregs activity score but shows a less positive correlation with other cells, such as CD8 T cells and NK cells (Fig. 7B). In addition, the KTRG risk score shows a negative correlation with several immune cell fractions, including monocytes, NK cells resting, and M1/M2 macrophages (Figure S9A). Besides Tregs, the risk score also shows a low positive correlation with the fraction of CD8 T cells and T cells CD4 memory activated. In addition, the immune checkpoint, CTLA4 and PDCD1 (PD1) are highly expressed in the high-risk group, but CD274 (PDL1) is highly expressed in the low-risk group (Figure S9B). The risk score is highly positively correlated with mRNA expression of PDCD1 and CTLA4 but negatively correlated with CD274 (Figure S9C). All in all, the relationship between the KTRG risk score and other immune cells is extremely complex, and we can not predict the potential immune therapeutic response among these results. Therefore, we conducted immune therapeutic response prediction by TIDE analysis. Notably, we found that most non-responders were distributed in the high-risk group, whereas most of the respondents were located in the low-risk group (Figure S9D). In addition, the risk score is higher in no-responders than that in responders (Figure S9E). From this humble evidence, we could speculate that the KTRG risk score could act as a potential predictor for cancer patients who receive immune therapy. We obtained two external immune therapy datasets (Alexandra and GSE78820). After merging the risk score based on their own KTRGs mRNA expression profile and clinical variables (such as survival data and immune therapeutic response data), the subsequent analyses were further performed. Similar to the results above, the high-risk group showed a low survival probability in these two datasets (Fig. 8A and E). Risk score also shows high predicted accuracy (for the Alexandra cohort, AUC at one year = 0.6410, AUC at three years = 0.7868; for the GSE78820 cohort, AUC at one year = 0.7072, AUC at three years = 0.7229) (Fig. 8B and F). Furthermore, the patients that prove to be non-responders (SD/PD) show a higher KTRGs risk score (Fig. 8C and G). In the same way, the patients in the high-risk group primarily harboured in the no-responder group, while those in the low-risk group were mainly distributed in the responder (CR/PR) group (Fig. 8D and H). These results provide potential evidence for our hypothesis that our KTRG risk score could be employed to act as a predictor for predicting immune therapeutic outcomes.
Fig. 7
Correlation between KTRGs and immune system. A The boxplot shows the difference in immune cell fraction and activity score in high and low KTRG risk groups. B The heatmap shows a correlation between mRNA expression of KTRGs and immune cell activity scores. *P-value < 0.05; **,P-value < 0.01; ***,P-value < 0.001; ****P-value < 0.0001
Fig. 8
Immune therapeutic response prediction. A and E Kaplain-Meier survival curve reveals the survival difference in the high KTRGs risk group in two immune therapeutic cohorts. B and F Time-dependent ROC curve of KTRGs risk score on predicting 1-/3-year survival. C and G KTRGs risk score difference between CR/PR and SD/PD cohorts in two immune therapeutic cohorts. D and H The distribution of high and low-risk groups in CR/PR and SD/PD cohorts in two immune therapeutic cohorts. *,P-value < 0.05; ***,P-value < 0.001; ****,P-value < 0.0001
留言 (0)