
記住我
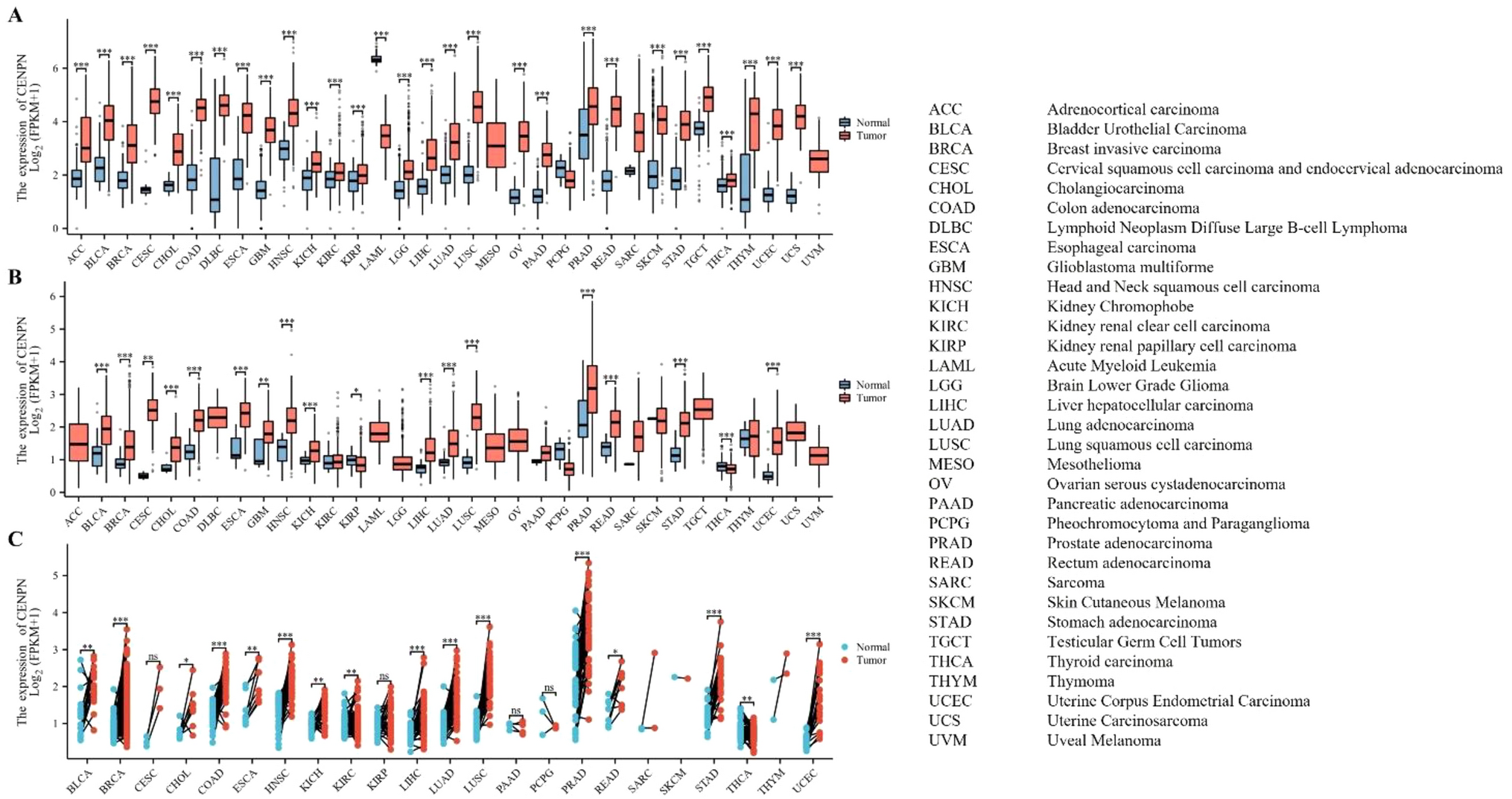

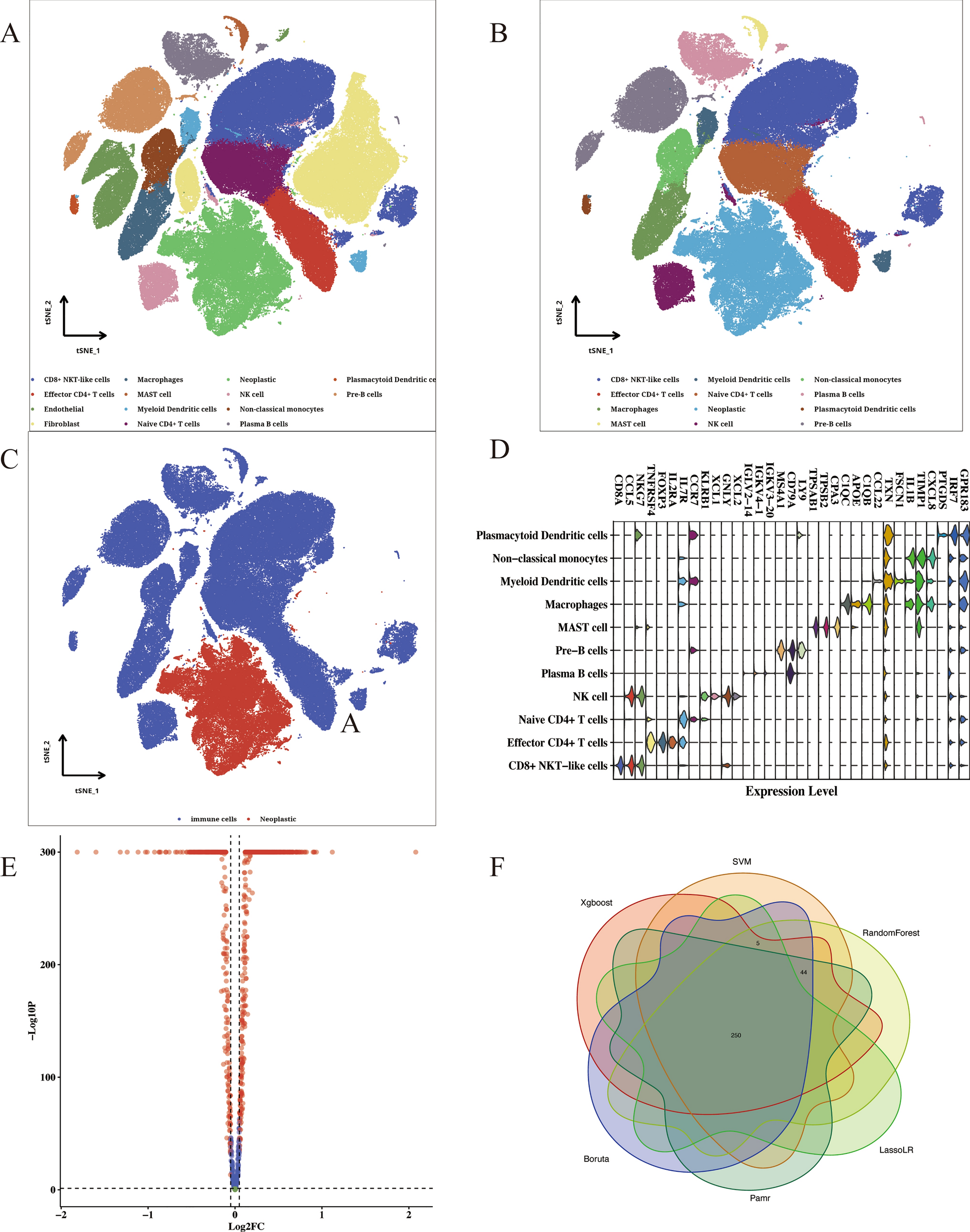


Performing dimension clustering analysis on the scRNA-seq dataset of ESCC, we identified all cell clusters in Fig. 1A. In the Fig. 1B, we selected 13 immune cell types for further analysis. Total 2,698 RNAs were defined as potential immune-related RNAs. 1,107 iuRNAs were were further identified when TSI < 0.45. We annotated marker gene with significant expression level in each immune cell (Fig. 1C). As presented in Fig. 1D, the distribution of immune cells and neoplastic cells was visualized. In addition, we identified DEGs between ESCC cells and immune cells in Fig. 1E. 299 significantly up-regulated DEGs were considered as TIIC-RNAs. After the integration of 6 machine learning algorithms, we finally obtained 250 valuable TIIC-RNA in Fig. 1F.
Fig. 1
Identification of TIIC-RNA at the single-cell level. A t-SNE plot of cell clusters in tumor microenvironment. B t-SNE plot of immune cells types. C Diagram of the expression level of marker genes in each immune cell. D t-SNE plot of the distribution of immune cells and neoplastic cells. E Volcano plot of DEGs screen by immune cells and neoplastic cells. F Venn map of most valuable TIIC-RNAs selected by 6 algorithms
3.2 Construction of TIIC signature scoreWe performed a univariate Cox regression analysis to investigate the prognostic value of TIIC-RNA for OS of ESCC patients. As it turned out, 12 TIIC-RNAs were identified in the TCGA dataset, including DNTTIP2, BIRC2, SQSTM1, ATP6V0E1, DNM2, TMED5, CAP1, VOPP1, NUP98, PPP3CC, RELB, and HLA-E (Fig. 2A). Three machine learning algorithms was conducted, containing CoxBoost (Fig. 2B), LassoCox (Fig. 2C, D), and Random Forest (Fig. 2E, F). We further determined three prognostic TIIC-RNA (Fig. 2G). We constructed the most reliable model based on a comprehensive C-index of externally validated datasets. TIIC signature score was developed based on prognostic TIIC-RNAs. In the TCGA-ESCC, GSE53622, GSE53624, and GSE53625 datasets, ESCC patients with high TIIC signature scores showed poorer survival outcomes in Fig. 2H. 1, 2, and 3 year- OS of ESCC patients were quantified by AUC values in TCGA-ESCC (0.652, 0.803, 0.729), GSE53622 (0.532, 0.631, 0.630), GSE53624 (0.507, 0.550, 0.562), and GSE53625 (0.515, 0.575, 0.586) (Fig. 2I).
Fig. 2
Construction of TIIC signature score. A Circle plot of 12 TIIC-RNAs by univariate Cox regression analysis. B Line map of 12 TIIC-RNAs evaluated by CoxBoost algorithm. C, D Diagrams of 12 TIIC-RNAs evaluated by LassoCox algorithm. E, F Diagrams of 12 TIIC-RNAs evaluated by Random Forest algorithm. G Venn plot of prognostic genes interacted by three algorithms. H K-M curves of OS of ESCC patients with high-TIIC and low-TIIC signature scores in different datasets. I ROC curves of 1, 2, 3 years of OS of ESCC patients with high-TIIC and low-TIIC signature scores in different datasets
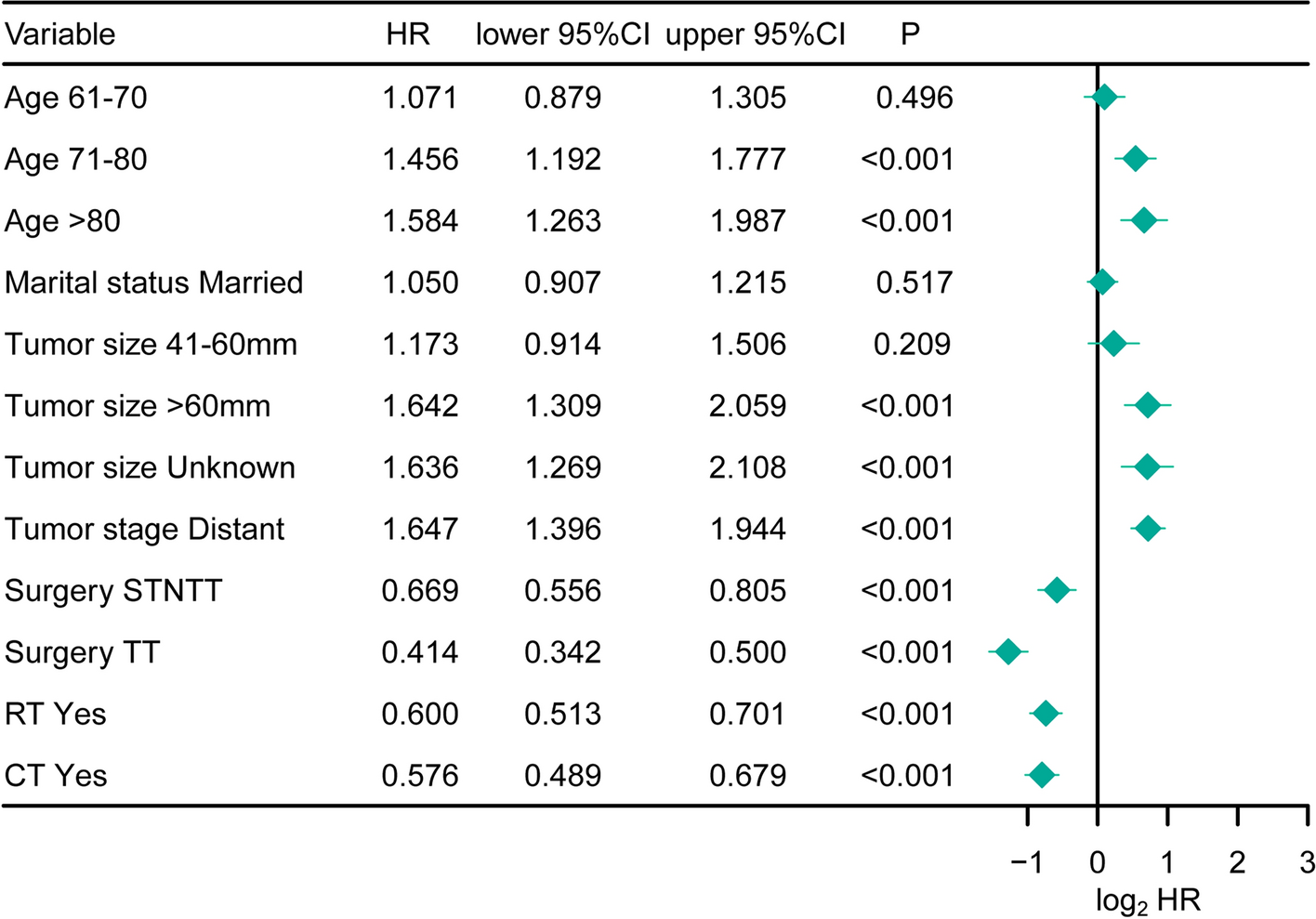
3.3 Prognostic value of TIIC signature score compared with clinical featuresIn the TCGA dataset, TIIC signature score was significantly correlated with survival status (p = 0.032), but not with tumor stage and TNM staging system (Fig. 3A). In addition, in the TCGA dataset, TIIC signature score showed better performance in terms of age, sex, tumor stage, and TNM staging system with high C-index (Fig. 3B). To further validate the prognostic performance of TIIC signature score, we included 30 published prognostic models and compared the C-index in the TCGA-ESCC, GSE53622, GSE53624, and GSE53625 datasets (Fig. 3C-F). Our TIIC model shows better performance in TCGA-ESCC, GSE53622, GSE53624, and GSE53625 datasets than most other published models.
Fig. 3
Prognostic value of TIIC signature score compared with clinical features. A Circos plot of the correlation of clinical characteristics between TIIC signature score groups. B Bar chart of the C-index of TIIC model and other clinical features in TCGA-ESCC, GSE53622, GSE53624 and GSE53625 datasets. C–F Line charts of the comparation between TIIC signature score and 30 published models in TCGA-ESCC, GSE53622, GSE53624 and GSE53625 datasets
3.4 Predicting biological mechanisms associated with TIIC signature scoreTIIC signature score is strongly positively correlated with several biological pathways, mainly including KEGG_LYSOSOME, KEGG_GALACTOSE_METABOLISM, and KEGG_PEROXISOME (Fig. 4A). We selected 8 pathways from the GO-BP and KEGG databases that were significantly different between the two scoring groups and showed the ssGSEA scores corresponding to the pathways (Fig. 4B). We showed the enrichment results of up-regulated genes in the high-TIIC group on Metascape, which showed that they were related to immune response and inflammatory response (Fig. 4C). We exhibited GSEA results for key genes as follows: In the high-TIIC group, key genes are correlated with CELLULAR_RESPONSE_TO_OXYGEN_CONTAINING_COMPOUND, REGULATION_OF_HYDROLASE_ACTIVITY, RESPONSE_TO_NITROGEN_COMPOUND, Response_oxygen_containing_compound, and RESPONSE_TO_OXYGEN_CONTAINING_COMPOUND. In the low-TIIC group, key genes are related to MOLTING_CYCLE, EPIDERMIS_DEVELOPMENT, CYTOKINE_PRODUCTION, and POSITIVE_REGULATION_OF_CELL_DEATH (Fig. 4D).
Fig. 4
Biological characteristics of TIIC signature score. A Heat map of GSVA analysis in TIIC signature score groups based on MsigDB. B t-SNE plots of differences of GO and KEGG pathway between the TIIC scoring groups. C Enrichment analysis of differentially expressed genes in high-TIIC group. D GSEA graphs of GO and KEGG for high- and low-TIIC groups
3.5 Correlation of TIIC signature score with immune-related featuresAs shown in Fig. 5A, we found that the activity of most immune cells decreased with the increase of TIIC score. At the same time, TIIC signature score showed difference in the expression levels of CD80, C10orf54, CD276, CD70 and methylation levels of CD80, CD28, CD274, CXCL1. There was also significant different correlation with the degree of CNV amplification of the immune checkpoint genes such as VTCN1, TNFSF9, and TNF, and CNV deletion of the immune checkpoint genes such as CD80, ICOSLG, PDCD1LG2, and CD274 (Fig. 5B).
Fig. 5
Correlation of TIIC signature score with immune-related features. A Heat map of correlation between TIIC signature score and immune infiltrated cells. B Heat map of correlation between TIIC signature score and immunomodulatory genes
3.6 Verification of the prognostic value of TIIC signature score for immunotherapy responseIn the IMvigor dataset, patients with low TIIC feature scores showed better survival outcomes (p = 0.02, Fig. 6A) and both responded against PD-L1 immunotherapy (Fig. 6B). The same results were validated in the Braun dataset (p = 0.0098, Fig. 6C) with no difference in response to PD-L1 immunotherapy (Fig. 6D). However, in the Nathanson and GSE78220 datasets, survival outcomes and response to PD-L1 immunotherapy showed no significant different in both groups (Fig. 6E-H). In addition, patients with low TIIC signature score responded better to immunotherapy in GSE165252 (Fig. 6I), GSE103668 (Fig. 6J), and GSE179351 (Fig. 6K), while patients with high TIIC signature score responded better to immunotherapy in GSE91061 (Fig. 6L), GSE35640 (Fig. 6M), and GSE126044 (Fig. 6N). The TIDE results showed that in the TCGA dataset, a low proportion of patients responded in the low TIIC signature score group (p = 0.04, Fig. 6O).
Fig. 6
Verification of the prognostic value of TIIC signature score for immunotherapy response. A, C, E, G K-M curve of OS of ESCC patients in the IMvigor, Braun, Nathanson, and GSE78220 between TIIC signature score groups. B, D, F, H Comparison of response to immunotherapy of ESCC patients in the IMvigor, Braun, Nathanson, and GSE78220. I–N Comparison of response to immunotherapy of ESCC patients in the GSE165252, GSE103668, GSE179351, GSE91061, GSE35640, and GSE126044. O The percent weight of responder and non-responder between TIIC signature score groups
3.7 Predicting metabolic characteristics associated with TIIC signature scoreTo investigate a wide range of metabolic characteristics in the two TIIC signature score groups, GSVA was performed against metabolic pathways in the KEGG database. TIIC signature score was significantly correlated with many metabolic pathways (Fig. 7A). It is worth noting that pentose phosphate pathway, fructose and mannose metabolism, galactose metabolism and glycosaminoglycan degradation activation rates were significantly higher in the high TIIC signature score group compared to low TIIC signature score group (Fig. 7B). In addition, TIIC signature score was positively associated with oxidative phosphorylation, other glycan degradation, primary bile acid biosynthesis and thiamine metabolism (Fig. 7C).
Fig. 7
Metabolic characteristics of TIIC signature score in TCGA dataset. A Heat map of GSVA analysis of 11 metabolic pathway subgroups between two TIIC scoring groups based on KEGG. B Maps of differences in metabolic pathways between the two TIIC scoring groups. C Scatter diagram of correlation between TIIC signature score and metabolic pathways
3.8 SNV mutation analysis and CNV analysisDifferent frequencies of chromosomal changes were observed in the two TIIC score groups (Fig. 8A). The waterfall map shows the mutations of top 50 genes in both groups. It can be seen that TP53 (90%), TTN (32.7%) and NFE2L2 (17.5%) are genes with high mutation rates, among which ADGRV1 (6.2%) and HTT (6.2%) have significant differences between the two groups (Fig. 8B). There is no statistically significant change of fraction genome altered (FGA), fraction genome gained (FGG), and fraction genome lost (FGL) between high- and low-TIIC signature score groups in Fig. 8C. CNV mutations in chr3 were significantly different between the two groups (p = 2.2e-06, Fig. 8D).
Fig. 8
SNV mutation analysis and CNV analysis. A Diagram of chromosomal changes based on GISTIC 2.0 in two TIIC signature score groups. B Waterfall map of genomic mutation landscape in two TIIC signature score groups. C Diagram of changes of FGA, FGG, and FGL in both TIIC signature score groups. D Diagram of CNV mutations in chr3 between two TIIC signature score groups
3.9 SMR and MR analysisGWAS data for ESCC were visualized using the Manhattan chart (Fig. 9A). Gene colocalization analysis was performed, showing two prognostic model genes (ATP6V0E1 and BIRC2) in Fig. 9B, C. MR analysis found that gastro-oesophageal reflux were not significantly associated with ESCC based on SNP of TIIC-RNA gene, but rs148710154 and rs75146099 SNP sites had a significant correlation between them (the result of a single SNP can be seen in gastro-oesophageal reflux_to_cancer_MR_snps_result.xlsx file, Fig. 9D-F).
Fig. 9
SMR and MR analysis. A Manhattan map of GWAS data for ESCC. B, C Gene colocalization analysis of two prognostic model genes (ATP6V0E1, BIRC2). D–F Scatter plot, funnel plot, and forest plot of MR analysis between gastro-oesophageal reflux and ESCC
留言 (0)