
記住我
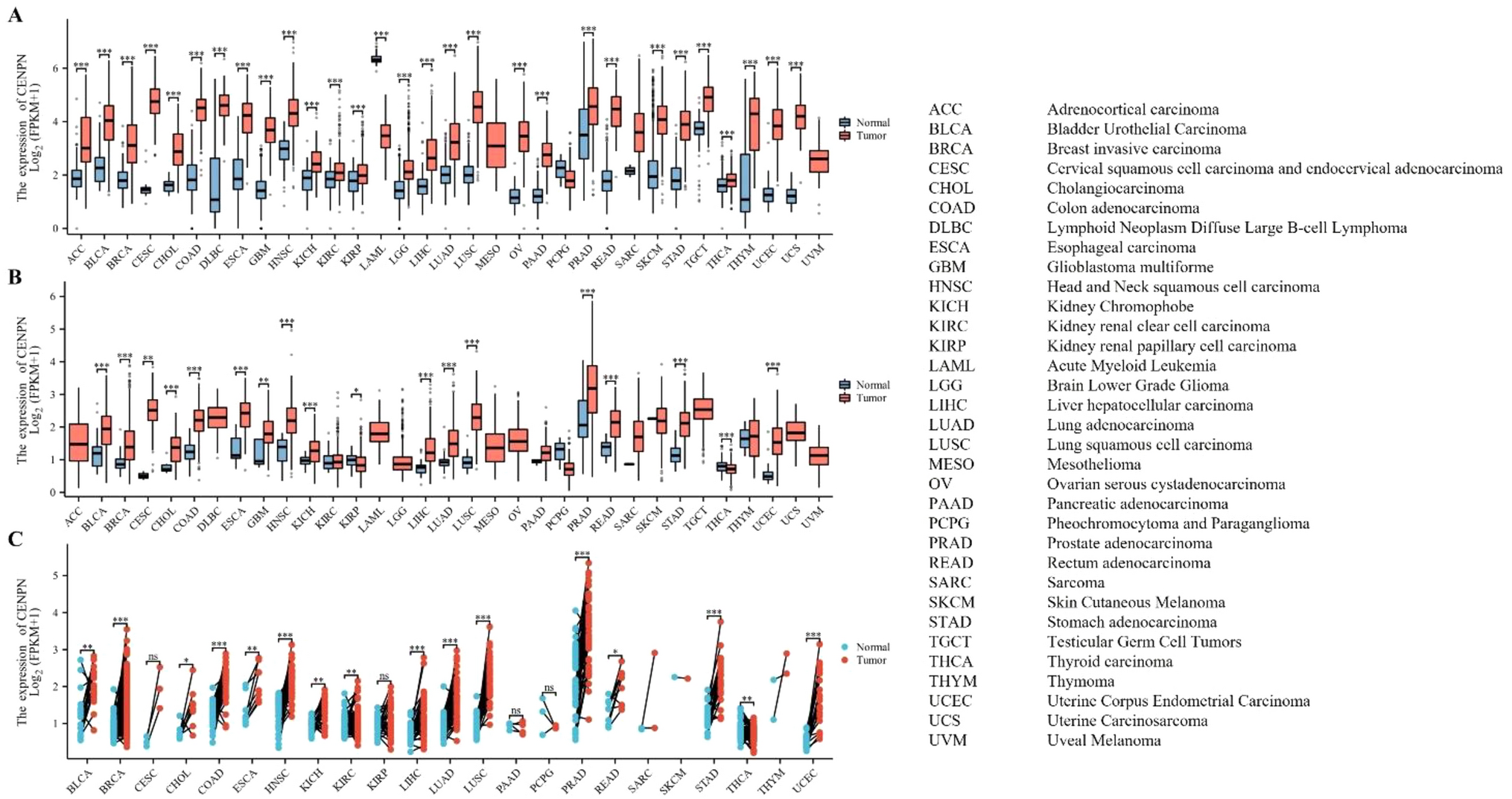


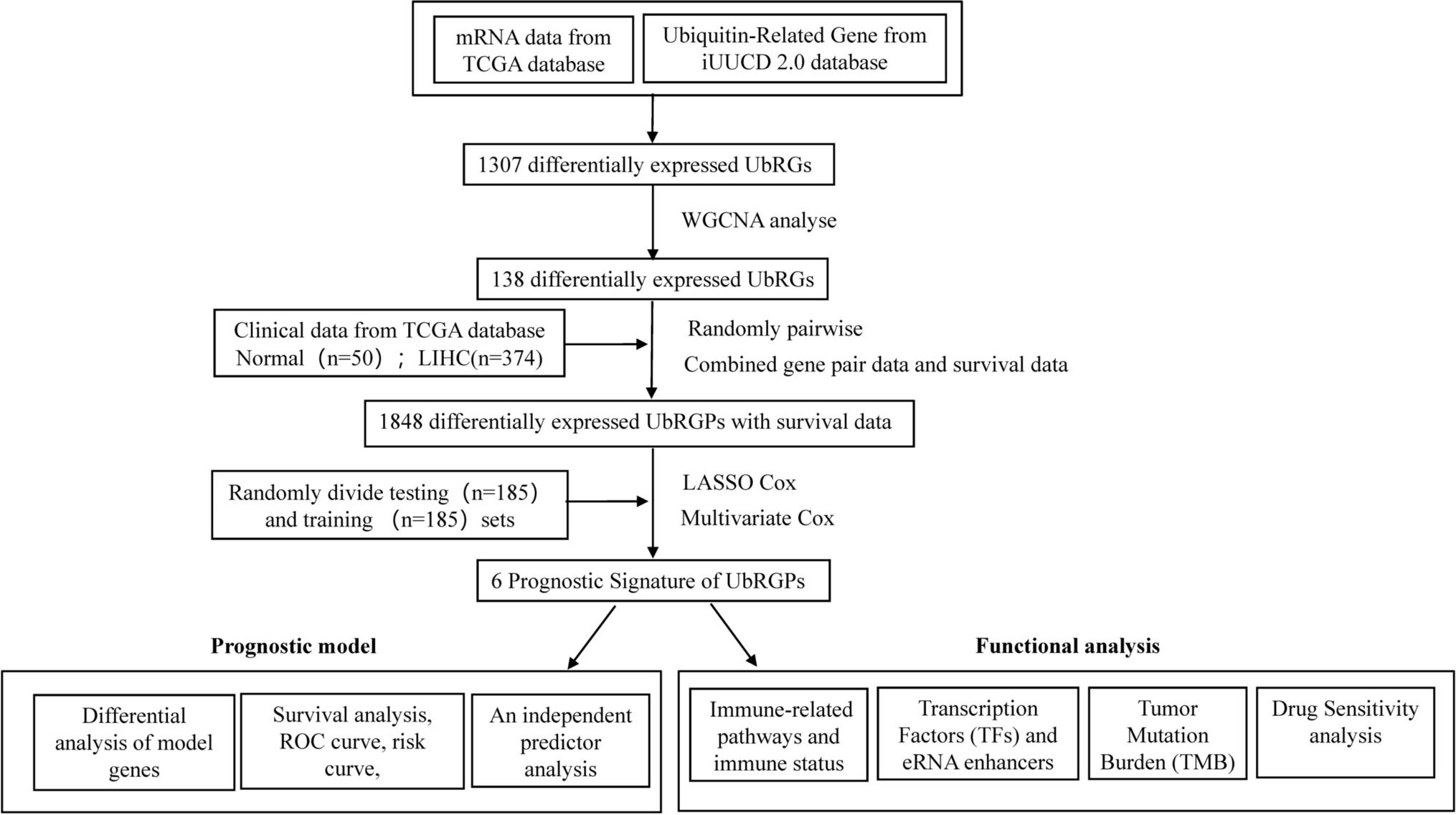

To construct the differentially expressed UbRGPs, we built a co-expression network of DEGs in 1307 UbRGs obtained from the TCGA-LIHC Transcriptome database and subsequently identified the co-expression genes of expression set modules that exhibit a significant correlation with clinical traits. A scale-free network was constructed using a soft threshold of β = 12 (R2 = 0.85) (Fig. 2A). All the genes were categorized into co-expression modules, each containing 50 distinct genes, and illustrated with different colors (Fig. 2B). We conducted plotting of the eigenvector clustering module (Fig. 2C) and gathered the MEbrown module that had the lowest p-value (Fig. 2D, p = 0.63*4E−48). Afterward, we assembled UBRGPs. Figure 2E shows the module membership in the brown module versus gene significance. Finally, the differentially expressed UbRGPs (0 or 1 frequency between 20 and 80%) were established for further analysis. To conclude, the differential expression of UbRGs was constructed to establish UBRGPs.
Fig. 2
WGCNA co-expression network analysis. A Analysis of the scale-free index and the mean connectivity for different soft-threshold powers. B Dendrogram of all differentially expressed genes clustered according to the measurement of dissimilarity. The color band illustrates the outcomes of the automatic single-block analysis. C Clustering of modular eigenvectors. D Correlation between modules and clinical trait according to Pearson correlation. E The module membership in brown module versus gene significance
3.2 Construction of the prognostic signature of UbRGPs in LIHCTo generate the prognostic signature of UbRGPs in LIHC, a prognostic model was formed by integrating the previously mentioned UBRGPs and TCGA-LIHC survival data and utilizing LASSO Cox regression analysis. The TCGA cohort was randomly divided into a testing set (n = 185) and a training set (n = 185). In the training set, thirty-six UbRGPs were found to be correlated with OS when the criterion of p < 0.001 was considered. The forest map (Fig. 3A) displays the 95% confidence interval (CI) and hazard ratio (HR) of each prognosis-related UbRGP, signifying that the majority of these gene pairs are risk and protective factors. Based on the optimal values of λ, six UbRGPs were chosen for the stepwise LASSO Cox analysis (Fig. 3B, C). In the end, six UbRGPs were utilized in the construction of the prognostic signature, and the particulars of these gene pairs can be found in Supplementary Table S2. According to the forest map, the WRAP53|CCNF, ESR1|RNFT2, FBXL6|DCAF13, THOC6|CDC20, and FBXW4|SHKBP1 were identified as protective factors with HR < 1, while UBE2T|NSMCE2 was the only risk factor with HR > 1 (Fig. 3, p < 0.05). Through differential analysis of model genes, it was found that the genes WRAP53, CCNF, UBE2T, NSMCE2, RNFT2, FBXL6, DCAF13, FBXW4, SHKBP1, THOC6, and CDC20 were upregulated in tumor samples, while ESR1 was downregulated in tumor samples compared to normal samples (Fig. 3D, E, p < 0.001). In summary, we constructed the prognostic signature of UbRGPs in LIHC and observed significant differences between LIHC and normal tissue.
Fig. 3
Construction of the Prognostic Signature of UbRGPs. A Prognosis-related UbRGs. B, C LASSO Cox analysis. D, E Differential analysis of model genes. F–I Kaplan–Meier plot of the risk score and OS, PFS. J ROC curves. K–M A scatter diagram of the risk score and survival status distribution. *p < 0.05; **p < 0.01; ***p < 0.001
The risk score was calculated using the multivariate Cox proportional hazard model.The train set was divided into high-risk group (n = 91) and low-risk group (n = 94) based on the median risk score, whereas the test set was classified into high-risk group (n = 97) and low-risk group (n = 88). The Kaplan–Meier curve consistently showed that patients in the high-risk group of OS and PFS had a significantly poorer prognosis than those in the low-risk group (Fig. 3F–I; F: All set, p < 0.001; G:Training set, p < 0.05; H:Testing set, p < 0.001; I: All set, p < 0.001). The prediction accuracy, as evaluated by AUCs, was reported to be 0.740, 0.707, and 0.645 in the 1-year, 3-year, and 5-year ROC curves, respectively (Fig. 3J). Additionally, the time-dependent AUC values were calculated (Supplement Fig. 8A). Figure 3K–M depicted a scatter diagram of the risk score and survival status distribution, as well as a heatmap of each patient in the training and testing sets (Fig. 3K–M; K: All set; L: Training set; M: Testing set). It can be inferred that high-risk patients had a higher probability of death earlier than low-risk patients. To conclude, the aforementioned findings demonstrate that the six UbRGPs signature can accurately predict the survival probability of patients with LIHC.
3.3 The risk scores were positively associated with poor pathological grading and clinical stage and served as an independent prognostic indicatorThe correlation between the signature-based risk scores and clinical parameters such as age, gender, immune score, cluster, and T, N, and TNM stages was examined in the TCGA cohort. Patients in stages Ш and stages II had a higher risk score compared to patients in stages I (Supplementary Figure S1A, p = 1.6E−05 and p = 0.0007). Additionally, patients in stages G4, G3, and G2 had a higher risk score than those in stage G1 (Supplementary Figure S1B, p = 0.00033, p = 5E−07, and p = 0.036), and patients in stages T4, T3, and T2 had a higher risk score than those in stage T1 (Supplementary Figure S1C, p = 0.00059, p = 0.00032, and p = 0.00017). However, there were no differences in terms of N stage, M stage, age, or gender (Supplementary Figure S1D–G, p > 0.05).
Univariate and multivariate Cox regression were analyzed to investigate the effect of the risk score. The results revealed that the risk score had a significant correlation with OS in the univariate Cox regression analysis (HR = 2.884, 95% CI = 1.967–4.229, p < 0.001, Supplementary Figure S1H). Furthermore, multivariate Cox regression analysis demonstrated that the risk score was an independent prognostic indicator (HR = 2.441, 95% CI = 1.580–3.770, p < 0.001, Supplementary Figure S1I). To evaluate the accuracy of the risk score prognostic indicator, the ROC curve was employed, which demonstrated that the risk AUC was 0.740. This outperformed other clinical features such as age, AUC = 0.531; gender, AUC = 0.509; grade, AUC = 0.499; stage, AUC = 0.671 (Supplementary Figure S1J). The accuracy of the risk score in predicting OS was high, as indicated by the results.
3.4 The clinical nomogram was established and demonstrated high accuracy in predicting OSTo accurately quantify LIHC, we developed a nomogram that integrates age, gender, stage, and risk score to predict OS. This nomogram enables a more precise assessment of patient risk, facilitating informed decisions regarding future treatment strategies (Fig. 4A). To evaluate the prediction accuracy of the nomogram, we employed a calibration curve that compares observed outcomes with predicted outcomes (C-index = 0.740, 95% CI: 0.687–0.793, Fig. 4B). The area under the curve (AUC) values at 1, 3, and 5 years were 0.804, 0.804, and 0.797, respectively (Fig. 4C), meanwhile we still calculate the time-dependent AUC values (Supplementary Fig. 8B). These findings indicate that the risk score serves as an independent prognostic indicator, and the nomogram effectively predicts the prognosis of LIHC patients. Notably, the AUC values in both the TCGA dataset and the validation dataset exceeded 0.7, demonstrating that the new signature has a significant capacity for predicting survival. In addition, compared with the nomogram model that includes risk scores, the nomogram model that only includes age, gender, grade, and stage shows lower accuracy in predicting OS (C-index = 0.690, 95% CI: 0.636–0.743, Supplementary Figure S2); AUC values at 1, 3, and 5 years were 0.755, 0.757, and 0.750, respectively (Supplementary Figure S2C), meanwhile we still calculate the time-dependent AUC values (Supplementary Fig. 8C). This novel signature shows potential as an independent prognostic factor and was utilized to evaluate the TCGA dataset of patients diagnosed with LIHC.
Fig. 4
Nomogram development and validation. A Nomogram to predict the 1-year, 3-year, and 5-year OS. B Calibration curve for the OS nomogram model in LIHC. C ROC curves
3.5 Internal functional mechanism investigation related to the prognostic signature3.5.1 The expression of TFs and eRNA enhancers related to the signature of UbRGsWe conducted an investigation into the association between the expression of the prognostic signature of UBRGs and TFs, as well as eRNA Enhancers. The expression of the prognostic signature of UBRGs was associated with 35 TFs, which include ZNF93, ZNF707, ZNF706, ZNF7, ZNF696, ZNF628, ZNF530, ZNF251, ZNF207, ZNF205, ZBTB17, ZBTB12, YBX1, THAP11, TFAP4, SAFB, MYPOP, MYBL2, MXD3, MTF2, MTERF3, MSANTD3, HSF1, FOXM1, E2F8, E2F7, E2F6, E2F5, E2F4, E2F2, DNTTIP1, DNMT1, CXXC1, CENPA, and ESR1 (Supplementary Figure S2A). Additionally, the prognostic signature of UBRGs was linked to 26 eRNA enhancers, which include ZNF337-AS1, THUMPD3-AS1, TENM3-AS1, STX4, STK3, STEAP1B, RNF139-AS1, PCAT1, NUTM2A-AS1, NBDY, MSH6, LINC01615, LINC00205, FOXO3B, FAM225A, FAM120AOS, EMG1, DCP1A, CHST12, CDKN2B-AS1, CDK6-AS1, AL355574.1, AF279873.3, AC069360.1, AC023403.1, and AC012574.2 (Supplementary Figure S2B).
3.5.2 Functional enrichment analysis related to the prognostic signatureTo elucidate the biological pathways associated with the prognostic signature, KEGG pathway analyses were conducted on the high-risk and low-risk groups. We investigated the disparities in biological functions between the two risk groups using Functional Enrichment Analysis. The results of the analysis revealed that the high-risk group exhibited enrichment in various biological processes, including the cell cycle, DNA replication, and hematopoietic cell lineage (Supplementary Figure S3A). In contrast, multiple metabolic-related pathways, such as fatty acid metabolism and drug metabolism via cytochrome P450, were found to be active in the low-risk group (Supplementary Figure S3B). These findings highlight distinct pathophysiological mechanisms underlying LIHC and suggest that this complexity may contribute to the differences in disease progression and patient outcomes.
3.5.3 Immune landscapes related to the signatureSubsequently, to investigate the immune landscape associated with the signature, we performed an analysis of TME cell infiltration characteristics. The tumor infiltrating immune cell heatmap revealed the presence of immune cells in each tumor sample across the two risk groups, which could be observed in Fig. 5A. Differences in the infiltration of certain immune cells between the high-risk and low-risk groups were observed, as illustrated in Fig. 5B. In the high-risk group, there was a lower infiltration of T cells CD4 memory resting, NK cells activated, monocytes, and mast cells resting compared to the low-risk group (Fig. 5B, p < 0.001, p = 0.036, p = 0.006 and p = 0.025). However, there was an increased infiltration of T cells CD4 memory activated and T cells follicular helper in the high-risk group (Fig. 5B, p < 0.001 and p = 0.032). To delve deeper into the correlation between risk scores and immune status, we carried out a differential analysis of immune-related functions and immune checkpoints. The results for B cells, mast cells, neutrophils, Tfh, NK cells, parainflammation, Type_I_IFN_Response, and Type_II_IFN_Response showed a significant difference between the high-risk and low-risk groups (Fig. 5C, p < 0.05). Apart from Tfh, the high-risk group showed lower levels of the other immune-related functions (Fig. 5C, p < 0.05). Significant differences in gene expression of checkpoint molecules, such as CD28, ICOS, TNFRSF9, HAVCR2, and LAG3, were observed between the high-risk and low-risk groups, with higher gene expression in the high-risk group (Fig. 5D, p < 0.05, p < 0.01, and p < 0.001). The results showed clear signs of immunocyte infiltration in two groups, and a correlation was observed between the tumor immune microenvironment and the risk score.
Fig. 5
Immune landscapes related to the prognostic signature. A, B Immune cell infiltration analysis in the two risk groups. C Immune-related function analysis in the two risk groups. D Expression level of immune checkpoints in the two risk groups. *p < 0.05; **p < 0.01; ***p < 0.001
3.5.4 TMB related to the prognostic signatureThe analysis of somatic mutations in both low-risk and high-risk groups revealed that the TMB of the high-risk group was significantly higher than that of the low-risk group (Fig. 6A, p = 0.05), and patients with high TMB had a shorter OS (Fig. 6B, p = 0.031). To conduct a survival analysis, we combined the risk score and TMB and determined that patients in the high-risk group with high TMB mutations had a dismal prognosis (Fig. 6C, p < 0.001). The outcome could potentially serve as a valuable prognostic indicator for LIHC.
Fig. 6
Analysis of TMB. A Comparison of TMB between the two groups. B Kaplan–Meier plot of TMB and OS. C Kaplan–Meier plot illustrating the relationship between OS and the risk score with TMB
3.6 Drug sensitivity analysis related to the prognostic signatureWe further explored the correlation between the sensitivity of 74 drugs of chemo- and targeted therapy and the prognostic signature. Patients in the high-risk group displayed reduced sensitivity to SB505124 (Supplementary Figure S4A, p = 2.5E−08), yet they exhibited reduced resistance to 72 other drugs, which included 5-Fluorouracil, Dihydrorotenone, I-BRD9, MK-1775, ML323, Oxaliplatin, Talazoparib, Vinblastine, and YK-4–279 (Supplementary Figure S4B-J, p = 4.5E−09, p = 1.7E−09, p = 1.8E−08, p = 1.7E−08, p = 1.6E−10, p = 2.5E−08, p = 4.3E−08, p = 4.3E−08, and p = 2.1E−09). Fig. S4A–J showed that these drugs are the top nine most significantly correlated drugs that impact prognosis. The results of sensitivity to other drug concentrations were shown in Fig. S4 (Supplementary Figure S4, p < 0.001). These findings imply that the prognostic signature has the potential to predict the sensitivity of chemotherapy and targeted therapy, thereby guiding clinical treatment choices and achieving satisfactory clinical outcomes.
3.7 The protein expression of UBRGs significantly increased in LIHCThe Human Protein Atlas was utilized to obtain immunohistochemistry-based antibody-specific staining scores in LIHC, which were classified into four grades: high, medium, low, and undetected (Supplementary Figure S5). The protein expression data correlated with the mRNA results (Fig. 3E), indicating that WRAP53, CCNF, NSMCE2, RNFT2, DCAF13, FBXW4, SHKBP1, THOC6, and CDC20 were highly expressed in LIHC tissues, while their expression was low in normal liver tissues (Supplementary Figure S5). The proteins of ESR1 were highly expressed in LIHC tissues, while the specific reasons for this result still require further exploration, as ESR1 mRNA expression was downregulated. As a result, the aforementioned UBRGs have the potential to serve as prognostic predictors or even treatment targets.
留言 (0)