To elucidate the function of lncRNA MIR210HG in hepatocellular carcinoma, we conducted various biological experiments to determine its mechanism of action, including CCK8 (Cell Counting Kit-8), western blot analysis, clone formation assay, the scratch wound healing assay, transwell migration and invasion assays, apoptosis assay, immunofluorescence assay and so on.
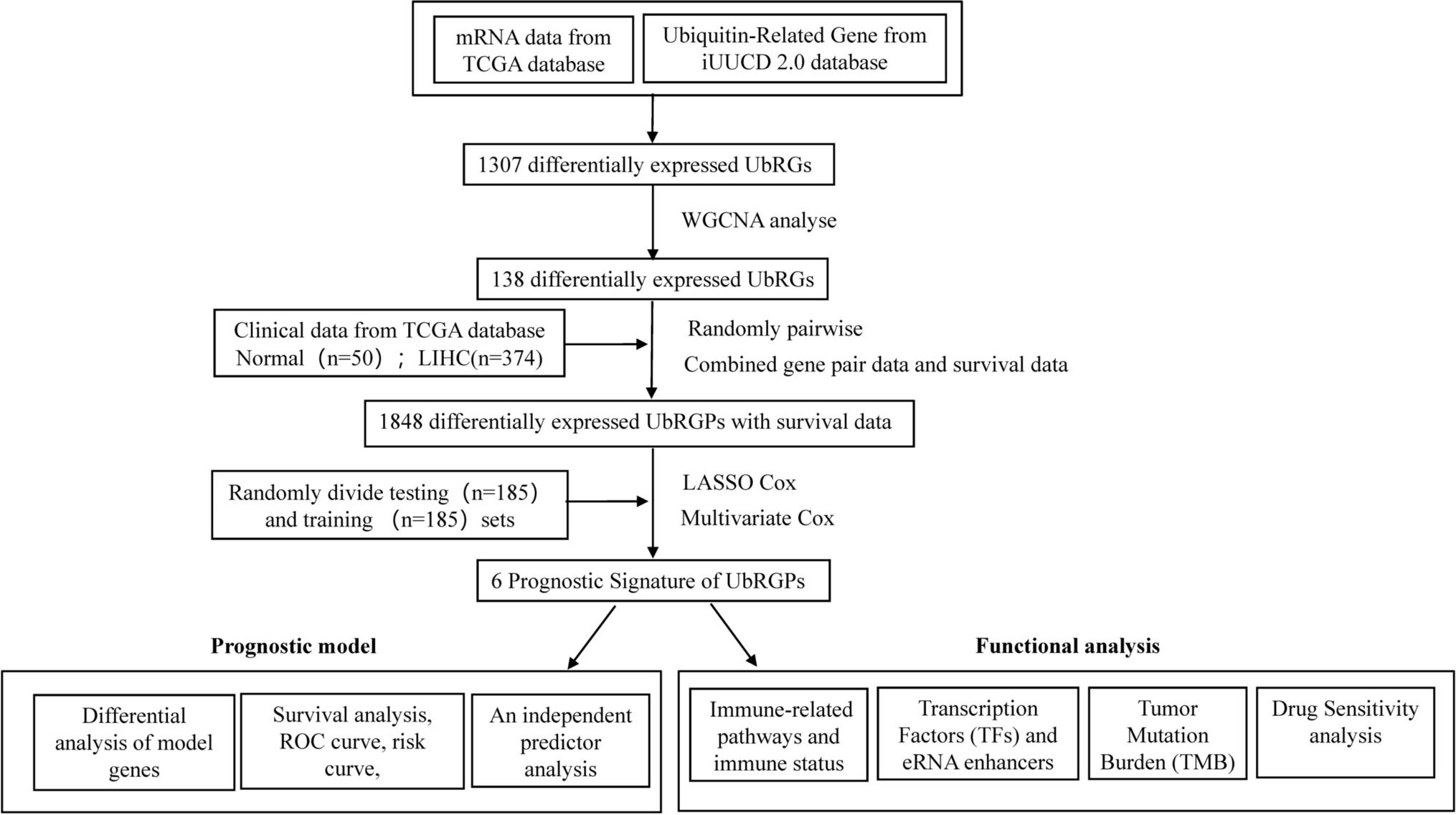
2.1 In silico and bioinformatics analysis: patient data acquisition
The Cancer Genome Atlas (TCGA) (https://www.cancer.gov/ccg/research/genome-sequencing/tcga) was used to acquire raw RNA-sequencing (RNA-seq) data and clinical information. Data Release 41.0:August 28,2024. The downloaded data was organized and pre-processed to enhance its adherence to the specified criteria and requirements.
2.2 Screening of autophagy-related lncRNAs
Relevant experiments were conducted to screen for lncRNAs associated with autophagy. LncRNAs linked to autophagy were selected using the criteria of a correlation coefficient exceeding 0.3 (R > 0.3), and a P value below 0.001 (P < 0.001).
2.3 Building the predictive signature
The univariate Cox regression model was employed to identify autophagy-associated lncRNAs whose expression levels were found to be significantly linked to the overall survival (OS) of the patient group diagnosed with hepatocellular carcinoma. Next, multivariate Cox regression analysis was performed on the lncRNAs associated with autophagy to determine their impact on patient survival as autonomous prognostic factors. Five specific lncRNAs associated with autophagy were ultimately identified through a hierarchical screening procedure and selected as potential candidates for prediction models.
2.4 Evaluation of the prognostic signature
Patients diagnosed with hepatocellular carcinoma were categorized into high-risk and low-risk groups based on their predictive risk scores, with the median risk score serving as a threshold. The OS of patients in high-risk and low-risk groups were compared by analyzing Kaplan–Meier survival curves and conducting two-sided log-rank tests. The predictive power of the prognostic signature in predicting patient survival outcomes was investigated using stratified survival analysis. Additionally, a multivariate and univariate Cox regression analysis was performed to assess the independence of the risk score from other clinical characteristics. Statistical significance was considered at a P-value less than 0.05.
2.5 Cell lines and culture
The Huh7 cell line, originating from human hepatocellular carcinoma, was obtained from the National Collection of Authenticated Cell Cultures. The Department of Pathogenic Biology at Hebei Medical University provided the human liver cancer cell lines Hep1 and Hep3B for research. The Chinese People's Liberation Army General Hospital provided LO2 and MHCC97-H. The cells were cultivated in DMEM medium supplemented with 10% fetal bovine serum; the incubator temperature was 37 °C with 5% CO2 concentration.
2.6 Cell transfection
GenePharma (Suzhou, China) designed and synthesized three specific siRNA sequences targeting lncRNA MIR210HG. Lipofectamine 3000 (Invitrogen, USA) was utilized to transfect cells. The sequence with the highest level of stability in the silencing effect was chosen for the subsequent experiment. The target sequences were as follows: si-MIR210HG: sense 5′- GAAAUAACCAAGCCGAGUUTT-3′ and antisense 5′- AACUCGGCUUGGUUAUUUCTT −3′.
2.7 Extraction of RNA and performing qRT-PCR
Total RNA extraction from cell lines was performed using Trizol reagent (Invitrogen, USA). The PrimeScript™ one-step qRT-PCR kit (TaKaRa, China) used reverse transcription to turn RNA into cDNAs. The 2−ΔΔCt method was employed to determine the variation in relative mRNA expression. GAPDH was used as an internal reference for normalization. The primer sequences were F-AATAACCAAGCCGAGTTGCCTCTG and R-TCTGGAGCACACAAAGGGAACAAG. The experiment was repeated three times to ensure the stability of the results.
2.8 Assessment of cellular growth
Cell proliferation was assessed using the CCK8 (Dojindo, Japan). Huh7 cells were transfected and then seeded into 96-well culture plates at a density of 5 × 103 cells/well. On days 1, 2, 3, 4, 5, and 6, the wells underwent a two-hour incubation period with CCK8 solutions, as per the manufacturer’s instructions. The absorbance at 450 nm was determined using a microplate reader. The cell proliferation percentages were calculated and adjusted to control values. The absorbance of transfected cells at the 450 nm wavelength was measured after rapamycin was added under the same conditions. The experiment was repeated three times to ensure the stability of the results.
2.9 Clone formation assay
After transfection, the cells were introduced into new 6-well plates (2 × 103 cells per well). The plates were incubated at 37 °C with 5% CO2 for 2–3 weeks. Incubation was terminated when colonies became perceptible to the naked eye. After the removal of the liquid above the sediment, the cells were rinsed two times with phosphate-buffered saline (PBS). The cells in 6-well plates were treated with paraformaldehyde solution for 20 mins, and the colonies were subsequently treated with 0.2% crystal violet solution for the same period. The wells were captured using a camera, and the Image J software was utilized to determine the quantity of clones in each well. The rates of clone formation in each well were subsequently computed. The colony formation rate of Huh7-transfected cells after adding rapamycin was measured under identical conditions. The experiment was repeated three times to ensure the stability of the results.
2.10 The scratch wound healing assay
The cells were cultured in 6-well culture plates at a density of 2 × 105 cells/well for 24 hours before transfection. To induce scratches, cells were scraped with a sterile gun tip after transfection. After removing the exfoliated cells, the remaining cells were cultured at a temperature of 37 °C in the presence of 5% CO2 in serum-free media. Photographs were obtained at 4 × magnification after 24 and 48 hours. Huh7 cells treated with rapamycin were photographed under the same conditions with an identical imaging microscope. The experiment was repeated three times to ensure the stability of the results.
2.11 Transwell migration and invasion assays
The migration and invasion assays were performed using Transwell chambers, either uncoated or coated with Matrigel (BD Biosciences, USA), as per the instructions provided by the manufacturer. The cells were placed in 0.2 mL of serum-free DMEM after a 48-hours transfection period for migration assays. A chemoattractant was added to the lower chamber using the complete medium. Cells were extracted from the upper membrane using cotton swabs after 48 hours. Subsequently, the lower membrane was then fixed for 20 mins with methanol and stained using 0.2% crystal violet. The invasion assay procedure was similar to the cell migration assay, with the exception that the Transwell membranes were pre-coated with Matrigel. Invasion and migration of Huh7-transfected cells were also measured following the addition of rapamycin. The experiment was repeated three times to ensure the stability of the results.
2.12 Apoptosis assay
After achieving a confluence of 70–80%, the Huh7 cells were seeded into 6-well plates and transfected for 24 hours using si-NC and si-MIR210HG. After centrifugation, 2 × 105 cells were washed with PBS. Annexin V/PI apoptosis kits were used to detect cell apoptosis (UE, Suzhou). Cellular apoptosis was measured using flow cytometry (BD Accuri C6 Plus) after applying YF 488-Annexin V and PI to the cells for 15 mins at room temperature in the absence of light. The experiment was repeated three times to ensure the stability of the results.
2.13 Immunofluorescence assay
Glass coverslips were used to seed the cell lines. Following a 5-min incubation in ice-cold methanol, the cells were rinsed three times with ice-cold PBS. Afterward, the cells were exposed to 1% BSA for 30 mins. Subsequently, antibodies targeting LC3B (ab51520, Abcam, UK) and P62 (ab56416, Abcam, UK) were applied and left overnight at a temperature of 4 °C. The cells were cultured for one hour at ambient temperature in the absence of light using goat anti-rabbit IgG (ab6721, Abcam, UK). Finally, the cells were incubated for 5 mins with DAPI, followed by visualization of the coverslips using fluorescence microscopy. The fluorescence intensity was analyzed in five locations selected at random using Images J. The experiment was repeated three times.
2.14 Western blot
The supernatant was extracted using RIPA (Radioimmunoprecipitation Assay) buffer (Solarbio, China) after the cells were collected. Proteins in tumor tissues were collected in a similar way. The concentration of protein was measured using a BCA (Bicinchoninic Acid) protein concentration assay kit (Solarbio, China). Subsequently, 20 µg of overall protein was added into an SDS-PAGE gel and the protein was moved to a PVDF membrane with a pore size of 0.45 µm. The membranes were immersed in 5% skim milk for 2 hours. The following antibodies for the experiments were utilized: LC3B (ab51520, Abcam, UK), P62 (ab56416, Abcam, UK), Bax (ab32503, Abcam, UK), BCL2 (ab182858, Abcam, UK), CyclinD1 (ab134175, Abcam, UK), mTOR (ab2732, Abcam, UK), p-mTOR (ab109268, Abcam, UK), Tubulin (ab7291, Abcam, UK), and GAPDH (ab181602, Abcam, UK). After the primary antibody had been incubated, the membrane was then incubated at 37 ℃ for 1 h with the appropriate secondary antibody. Color development and exposure were performed using the Protein Blot Detection System. The experiment was repeated three times to ensure the stability of the results.
2.15 Animals
The pathogen-free environment in which male BALB/c mice aged 4–6 weeks were raised was obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. For the blank control experiment, a cohort of five male mice was used. The number of mice in the negative control group and the experimental group was five. Phosphate buffer was used to suspend Huh7 cells that had undergone transfection with shRNA-NC or shRNA-MIR210HG. Subcutaneously, 5 × 106 cells/1 mL were injected into the left hind abdomen of mice. Tumor sizes were assessed every three days using vernier calipers. The animals were sacrificed 21 days after implantation. All operations were approved by the Laboratory Animal Ethical and Welfare Committee of Hebei Medical University (No. IACUC-Hebmu-2022037), and adhered to the relevant guidelines set by the National Research Council's Guide for the Care and Use of Laboratory Animals.
2.16 Statistical analysis
A total of three trials were conducted. The findings were displayed as the average ± SE. The differences between groups were compared using the GraphPad prism. The statistical significance was denoted as *P < 0.05, **P < 0.01, and ***P < 0.001.




留言 (0)