2.1 Data collection
The data analyzed in this study were from two microarray datasets, GSE12417 and GSE37642, sequenced on the GPL96 platform. The reason why we choose GSE12417 is for it contains163 samples of bone marrow or peripheral blood mononuclear cells from adult patients with untreated acute myeloid leukemia. And we choose GSE37642 is for it contains 562 samples (140 HGU-133plus2; 422 HGU-133A; 422 HGU-133B) from adult patients with acute myeloid leukemia (AML). They were obtained from the Gene Expression Omnibus database, including the gene expression profile and corresponding clinical information. GSE12417 is the training set. In addition, to verify the differences in FHL1 gene expression between normal cells and AML, another dataset of coordinated RNA sequencing FPKM including Genotype-Tissue Expression, The Cancer Genome Atlas, and Target Queue was obtained from UCSC Toil.
2.2 Screening and identification of prognostic genes
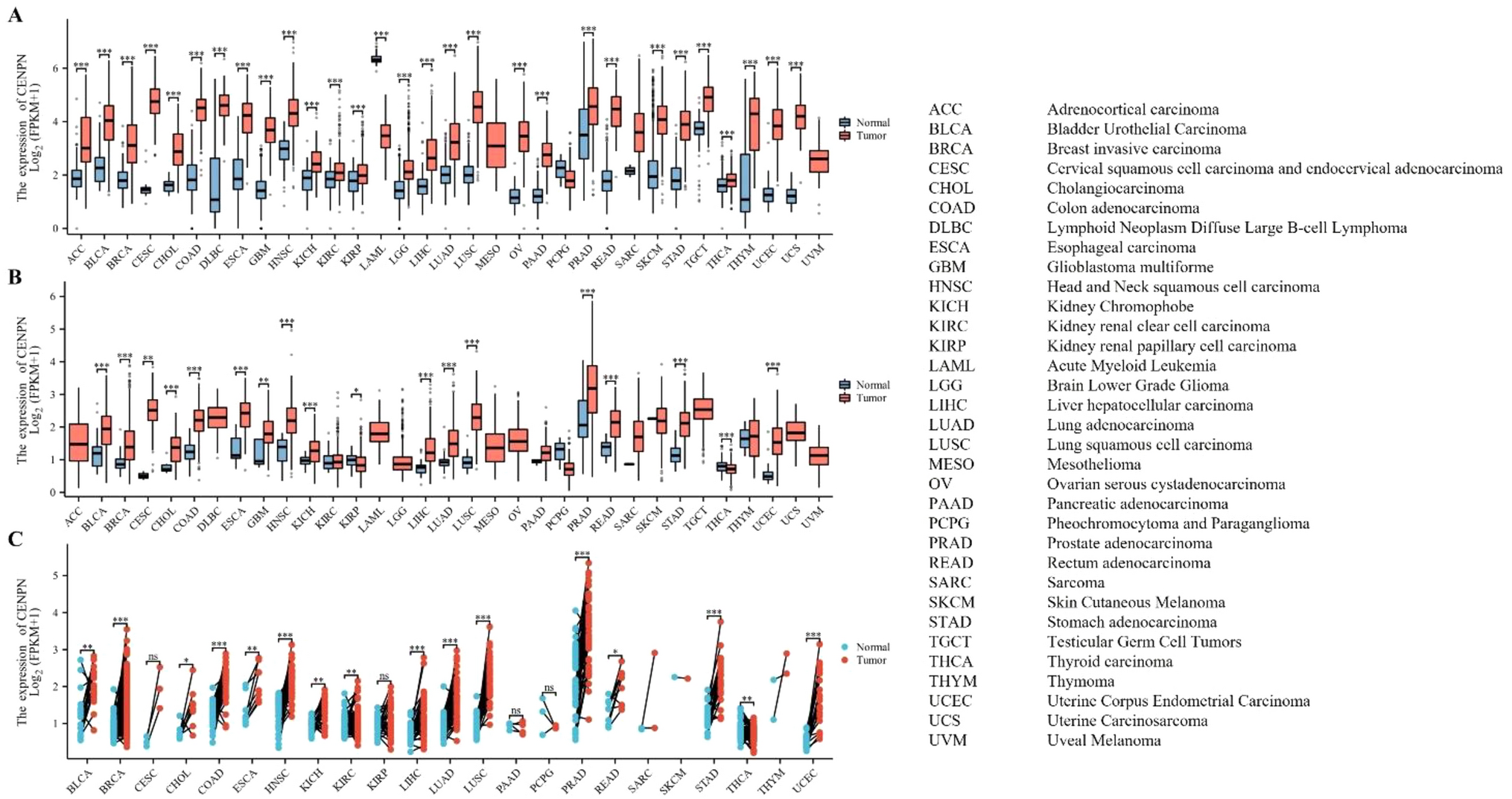
Kaplan–Meier survival analysis and univariate Cox regression analysis were used to screen all genes in the GSE12417 dataset. Based on P < 0.05 in both analyses, the gene with the most significant prognostic value (the largest area under the curve) was selected as the biomarker for subsequent analysis based on receiver operating characteristic analysis. We then assessed the clinical relevance of the biomarker. Based on French–American–British (FAB) classification and patient age, we created a box chart to visualize the expression distribution of biomarkers, and we evaluated the clinical correlation by univariate and multivariate Cox regression analysis. Kaplan–Meier survival analysis and clinical relevance assessment were validated with GSE37642.
2.3 Screening and functional identification of differentially expressed genes
The GSE12417 dataset was divided into high- and low-expression groups according to the median expression value of the biomarkers with the most significant prognosis. The R package limma was used for difference analysis between the two groups, with screening criteria of |log2 (fold change) |≥ 1.5 and false discovery rate < 0.05 [15]. Volcano plots and heat maps were used to visualize gene expression patterns. Then, we conducted Gene Ontology, Kyoto Encyclopedia of Genes and Genomes, and correlation analyses to find the action pathways of the differentially expressed genes and the correlation between them.
2.4 Gene set enrichment analysis
To reveal the potential pathway related to FHL1 expression, we used gene set enrichment analysis (GSEA) to identify the enrichment items in the GSE12417 dataset. GSEA generated an ordered list based on the correlation between all genes and FHL1 expression, indicating significant differences between high- and low-FHL1 groups [16], arranged 1000 times for each analysis. The enrichment pathways were sequenced with a P value and a normalized enrichment score. P < 0.1 was considered statistically significant.
2.5 Cell culture and gene silencing and overexpression
The M3 cell line HL60 from AML was used in this study. The cell lines were cultured in RPMI 1640 medium containing 10% fetal bovine serum at 37 °C and 5% CO2. Logarithmic growth cells were used for further experiments.
To determine whether there are differences in FHL1 expression between normal cells and AML, we first analyzed the expression values of FHL1 gene extracted from 337 whole blood data and 417 AML data obtained from UCSC Toil, then we used quantitative real-time PCR (qRT-PCR) and Western blot experiments to further verify the expression of FHL1 in neutrophils and HL60 cell lines.
First, HL60 cells were transfected with human FHL1-targeted small interfering RNA (siRNA) and negative control siRNA, designed, and synthesized by RiboBio Co. (Guangzhou, China) to silence the FHL1 gene. According to the manufacturer’s instructions, siRNA was transfected with 50 nM (1 × 105 cells/mL). Western blot was performed 48 h later to assess the transfection efficiency. The sequence of siRNA was as follows: si-FHL1: GGGAAGAAGTATGTGCAAA. Second, FHL1-overexpressed plasmid pcDNA3.1-FHL1 and pcDNA 3.1-vector (NC), constructed by Fenghui Biotech (Hunan, China), were transfected into HL60 cells with Lipofectamine 2000. Western blot was performed 48 h later to assess the transfection efficiency.
2.6 qRT-PCR and Western blot
The effects of silencing and overexpression were evaluated by qRT-PCR and Western blot. Total RNA was extracted from cultured cells with TRIzol and then reverse transcribed to complementary DNA in accordance with the manufacturer’s protocol (DP419; Beijing Tian Gen Biochemical Technology Co., Ltd., Beijing, China). The fluorescence intensity of TB Green in the reaction solution was detected, and the target gene was accurately quantified. β-Tubulin was used to standardize RNA expression. The results were calculated via the threshold cycle (2−ΔΔCt) method. Primers were designed and synthesized by Invitrogen (Shanghai, China), and the sequence was as follows: FHL1-human-qF 5′TCTGGCTCTGGAGCTAATTTGG3′;FHL1-human-qR5′TGGCAGTCAAACTTCTCCGC3′. In Western blot, the proteins were isolated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a 0.45-m polyvinylidene difluoride membrane. After the membrane was closed with 5% bovine serum albumin, the primary antibodies were used. Rabbit anti-FHL1 (1:1000; Proteintech, Wuhan, China) and rabbit anti-β-tubulin (1:5000; Abcam, Cambridge, UK) were incubated overnight at 4 °C. After washing the polyvinylidene difluoride membrane with Tris-buffered saline/Polysorbate 20 3 times (5 min/wash), we incubated the membrane with sheep anti-rabbit secondary antibody at room temperature for 2 h. Then the membrane was washed and developed to detect the protein expression level.
2.7 Detection of cell viability and apoptosis after treatment with cytosine arabinoside and daunorubicin
Cytarabine and daunorubicin were dissolved in dimethyl sulfoxide and stored at –20°, both purchased from Shenggong Bioengineering Technology Limited (Shanghai, China). HL60 cells with knockdown and overexpression of FHL1 were treated with cytosine arabinoside at 100 nM, 200 nM, and 500 nM and daunorubicin at 0.1 nM, 0.2 nM, 0.5 nM, 1 nM, and 2 nM, respectively, to determine whether expressions of the FHL1 gene in HL60 cell lines differed in drug sensitivity. Cell activity was detected by Cell Counting Kit-8 (CCK8). HL60 cells at a density of 104/100 µL were spread on a 96-well plate and treated with cytarabine and daunorubicin at different concentrations for 24 h. After 10 µL of CCK8 reagent was added to each cell, the cells were incubated for 4 h. The absorbance at 450 nm was measured with a microplate reader, and the cell viability was calculated. Cell viability (%) = (A Treatment group – A blank)/(A Control group – A blank) × 100. Cell apoptosis was detected with an Annexin V‐FITC/PI Apoptosis Kit (Sangon, China). HL60 cells at a density of 104/100 µL were inoculated into 24-well plates and treated with cytarabine and daunorubicin at different concentrations for 24 h. Annexin V-FITC/PI (5 μL) was added to 100 µL of cell suspension and incubated at room temperature in the dark for 10 min, then cell apoptosis was detected by flow cytometry.
2.8 Statistical analysis
R software version 3.6.1 was used for bioinformatic analysis, and GraphPad Prism version 8 was used to display the following experimental results. Wilcoxon rank-sum and Kruskal–Wallis tests were used to evaluate the differences between the groups. Unless otherwise specified, P < 0.05 was considered statistically significant.




留言 (0)