
記住我



Male C57BL/6 mice, 8 weeks old, were obtained from the Jackson Laboratory, Farmington, US. Furthermore, these animals were accommodated in polypropylene cages lined with wood shavings, with bedding changes occurring twice a week. Throughout the experiments, the animals were maintained in consistent environmental conditions (temperature: 22ºC ± 2ºC, with air exhaustion, and a 12 h light/12 h dark cycle). They had unrestricted access to water and standard chow diet ad libitum.
We confirm that all experimental protocols conducted in this study were approved by the Ethics of Animal Experiments Committee at the University of Virginia (Protocol number 4096). Additionally, we affirm that the reporting of all methods in this study aligns with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines, ensuring transparency and rigor in presenting our findings.
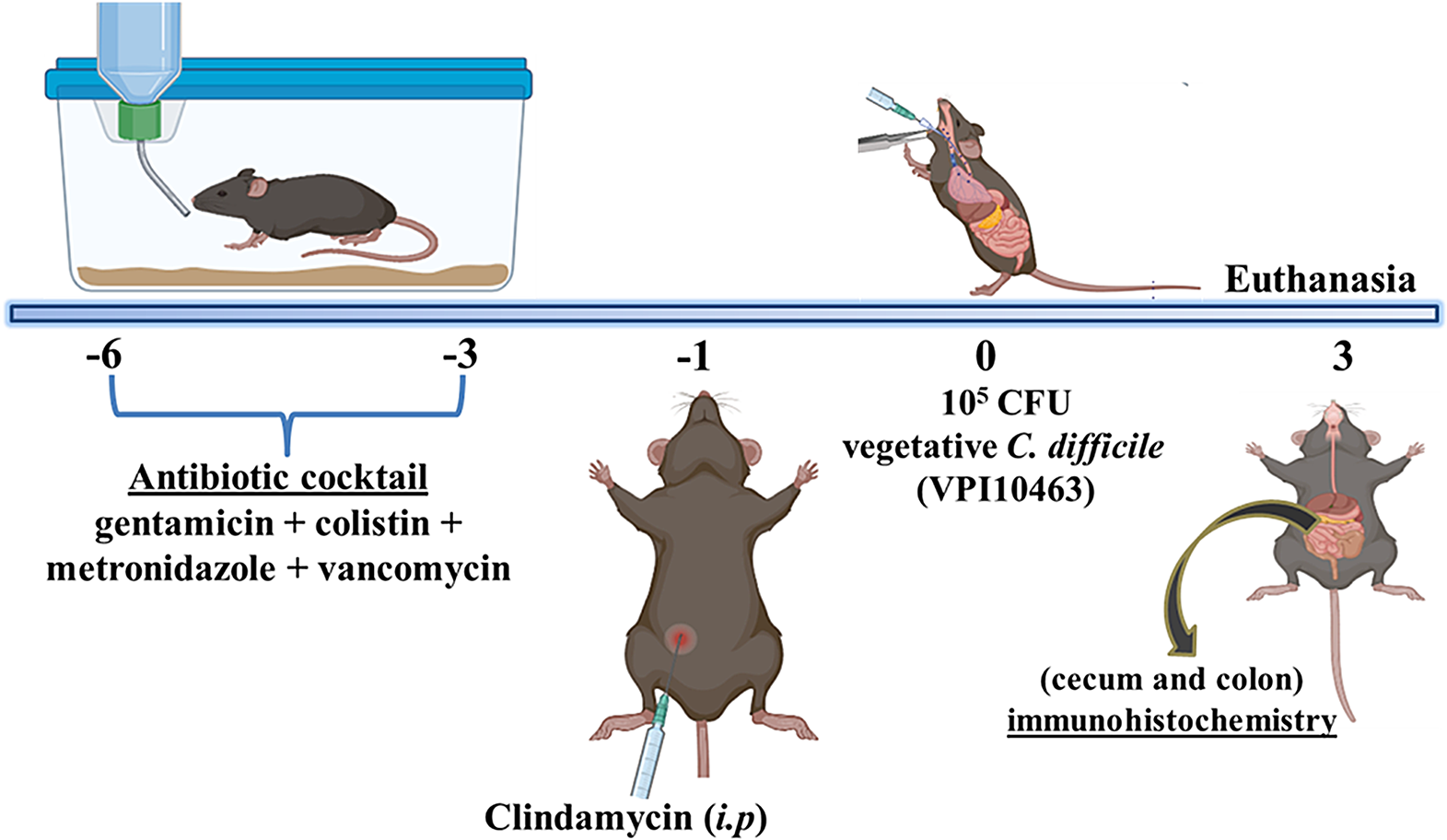
C. difficile infection modelThe murine CDI model utilized in this study follows a well-established protocol due to its ability to mimic clinical symptoms, such as severe diarrhea, presented by humans with CDI [8]. This model consists of disturbing the animal’s microbiota and thus facilitating colonization or infection by C. difficile in wild-type (WT) C57BL/6 mice (n = 6 for each group). The animals received a cocktail of antibiotics in their drinking water for 3 days, consisting of 0.035 mg per mL of gentamicin, 850 U per mL of colistin, 0.215 mg per mL of metronidazole, and 0.045 mg per mL of vancomycin, as illustrated in Fig. 1. Following a 2-day antibiotic-free period, the animals received an intraperitoneal injection of clindamycin (32 mg per kg, i.p.) 1 day before C. difficile challenge. Subsequently, 105 CFU (in 100µL of chopped meat broth, a pre-reduced medium) of the vegetative C. difficile strain VPI10463 (ATCC 43255, tcdA + tcdB + cdtB-) was administered by oral gavage. Control mice received chopped meat broth (100 µl). The mice’s weight and disease symptoms development were monitored daily. Three days after infection, animals were euthanized using a combination of ketamine (180 mg. kg; i.p) and xylazine (15 mg/kg, i.p.). Segments from the cecum and colon were collected and processed for immunohistochemistry analysis.
Fig. 1
C. difficile infection model induction scheme
ImmunohistochemistrySection (4 μm thick) were prepared from paraffin-embedded mouse cecum and colon tissues. After deparaffinization, antigens were recovered by incubating the slides in citrate buffer (pH 9.0) for 20 min at 95 °C in a PT link tank (DAKO). To reduce non-specific binding, endogenous peroxidase was blocked with 3% H2O2 for 10 min. The sections were then incubated overnight with an Anti-TRPV4 antibody (ab39260, Abcam, 1:1000), followed by a 30-minute incubation with polymer HRP (K8000, Dako). The antibody-binding sites were visualized by incubating the samples with diaminobenzidine–H2O2 (DAB, Dako) solution. The brown coloration indicates positive staining. The number of immunopositive cells was quantified using ImageJ software by counting cells with positive staining in ten distinct fields per slide, focusing on the “hot areas” (regions with intense staining). This analysis used one slide per animal and four specimens per group [21].
In vitro studiesRat EGC culture and treatmentThe immortalized EGC line used in this study comes from the jejunum region of Rattus norvegicus ATCC (PK060399egfr CRL-2690, Virginia-United States). These cells have demonstrated morphological and functional characteristics comparable to primary EGCs [22].
EGCs were cultured in DMEM medium (Dulbecco’s Modified Eagle’s Medium, Gibco) and supplemented with 10% fetal bovine serum, 1% antibiotics (100 mg/mL penicillin and 100 mg/mL streptomycin, Gibco), and 1 mM sodium pyruvate (Gibco) at 37 °C in a humidified incubator under 5% CO2 for no more than 25 passages. For all experiments in this research, EGCs were released from the culture flasks using 0.05% trypsin-EDTA for 5 min. The cells were incubated with the TRPV4-specific antagonist (RN-1734–100µM, Cayman, 946387-07-1) for 1 h before incubation with TcdA (50ng/mL) or TcdB (1ng/mL). In an additional experiment, we included a group of cells exposed simultaneously to both toxins. We maintained the same exposure time and concentrations evaluated in individual toxin treatments, with or without the TRPV4 antagonist. The concentration of RN-1734 was obtained from the results of the MTT assay (Fig. S1 – supplementary material). Furthermore, it is worth highlighting that TcdA and TcdB used in the study were purified from TechLab (Virginia-United States), and these, in turn, were produced by the C. difficile strain VPI10463.
Quantitative real-time PCR (qPCR)Initially, we aimed to determine whether EGCs express the TRPV4 receptor, establishing this as a prerequisite for advancing our research. To achieve this objective, we investigated the gene expression of TRPV4 by the Polymerase Chain Reaction (PCR) at 2, 12, and 18 h following incubation with either TcdA or TcdB. Subsequently, our research extended to examine other genes that may play a role in the pathophysiology of CDI, such as interleukin-6 (IL-6), interleukin-1 beta (IL-1-β IL-1β), and antiapoptotic B-cell lymphoma family gene 2 (bcl-2) genes, using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the reference gene.
EGCs (6 × 105 cells/well), at passage 20, were cultivated in 6-well plates and incubated with TcdA (50ng/mL) or TcdB (1ng/mL) for 18 h. The control group of cells was cultured exclusively in a culture medium (DMEM) without exposure to C. difficile toxins (control). After incubation, total RNA extraction was performed using a RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) on the QIAcube platform (Qiagen). Furthermore, RNA was quantified using the equipment Nanodrop. The RNA’s purity was evaluated using the following proportions: nucleic acids/proteins (260/280) and nucleic acids/other contaminants (260/230). Then, the RNA underwent reverse transcription using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol.
Amplification of TRPV4, GAPDH, IL-6, IL-1-β, and bcl-2 genes in cell samples was carried out using the StepOne apparatus. Amplification of specified genes in EGC samples was performed using a CFX Connect system (Bio-Rad) with the following conditions: 95 °C for 30 s, 40 cycles of 95 °C for 5 s, and 60 °C for 30 s, and analysis of the melting curve from 65 to 95 °C in increments of 0.5 °C for 2 s each. Gene expression was calculated by the Livak & Schmittgen method (2–ΔΔCt).
Following the same procedural steps described, the cells were co-incubated for 18 h with either TcdA or TcdB and the TRPV4 pharmacological modulator (RN-1734). We have also analyzed and compared the protein and gene expression profiles when cells were pre-treated with the TRPV4 modulator for one hour before toxin exposure. The primer sets used for these experiments are listed in Supplementary Material (Table S1).
Western blotting analysisTo assess the expression of TRPV4 protein, EGCs (6 × 105 cells/well) were seeded into 6-well plates and subsequently incubated with TcdA or TcdB for 18 h.
Following incubation, the supernatant was discarded, and the cells were lysed with RIPA lysis buffer (Thermo Fisher Scientific, containing EDTA and phosphatase-free protease inhibitor). The lysate was centrifuged for 17 min at 4 °C and 15.773 g, and the supernatant was collected for further analysis.
Protein concentrations were determined using the bicinchoninic acid assay following the manufacturer’s instructions (Thermo Fisher Scientific). Forty micrograms of protein, previously mixed with Laemmil sample buffer and β-mercaptoethanol, were denatured at 95 °C for 5 min. Subsequently, these proteins were separated on 10% BIS-Tris gel and transferred onto PVDF membranes for 2 h.
Following the transfer, the membranes were blocked with a 5% blocking solution (BioRad) at room temperature for 1 h. They were then incubated overnight at 4 °C with primary antibodies (anti-α-tubulin 1:500, Sigma-Aldrich T8203 and anti-TRPV4 1:2000, abcam39260). After primary antibody incubation, membranes were exposed to secondary antibodies (anti-mouse 1:500 and anti-rabbit 1:500) for 1 h and 30 min. Membranes were washed in Tris-buffered saline containing 0.05% Tween 20 (TSB-T) and then incubated with Enhanced Chemiluminescence – ECL (Biorad 1705060). The chemiluminescence signal was detected using a ChemiDoc system (BioRad). Densitometric quantification of bands was performed using ImageLab software (BioRad).
In a distinct experimental set, replicating all the previously described procedural steps, the protein expression of cleaved caspase-3 (an apoptosis marker) was assessed in EGCs exposed to TcdA and TcdB in the presence or absence of the TRPV4 antagonist. Specifically, cells were pretreated with RN-1734 (100µM) 1 h before toxin incubation. The antibodies used included primary antibodies (β-actin 1:200, Santa Cruz sc-7210; cleaved Caspase-3, 1:400, Millipore AB3623) and the secondary antibody (anti-rabbit; 1:400).
We used the ImageJ software to quantify Western blot bands. After selecting each band of interest by drawing a rectangular box around it, ensuring the band was fully encompassed, the software generated a peak for each band. The area under the curve of each peak corresponded to the band’s intensity. The signal intensity, expressed in pixels, was directly proportional to the target protein concentration. To account for any variations in sample loading, the intensity of each target band was normalized by dividing it by the intensity of the corresponding loading control band. This normalization step ensured standardized data and allowed for reliable comparisons between samples.
ImmunofluorescenceImmunofluorescence was performed to investigate the localization of the TRPV4 receptor in EGCs following an 18-hour treatment with TcdA or TcdB. EGCs at passage 17 were seeded in 24-well plates (4 × 104 cells/well). After the 18 h incubation period with the toxins, the cells were fixed in 4% paraformaldehyde (500 µL per well) for 15 min, followed by permeabilization in PBS containing 0.025% Triton X100 (Sigma-Aldrich) and 0.2% bovine serum albumin (BSA, Sigma-Aldrich) for 15 min. After permeabilization, cells were blocked with 0.25% Triton X100 (Sigma-Aldrich) and 5% BSA (Sigma-Aldrich) in PBS at room temperature for 1 h, then washed with a washing solution. Then, the cells were incubated with an anti-TRPV4 primary antibody (1:1000, abcam39260) or cleaved Caspase-3 (1:400, Millipore AB3623) at 4°C overnight. After three washes with wash buffer (0.01% Tween 20 in PBS), cells were overnight incubated with AlexaFluo 488 secondary antibody (1:400, Invitrogen). Following a wash with PBS, cells were mounted with ProLong Gold antifade reagent containing DAPI (Thermo Scientific, P36931). The samples were visualized by confocal fluorescence microscopy (LM10-Confocal Zeiss).
Immunofluorescence for TRPV4 or cleaved Caspase-3 was assessed using ten digital images captured in each section, with four samples per group. The average positive fluorescence intensity was quantified using Zeiss software. The average fluorescence intensity was quantified using Zeiss software. The software allows the selection of specific channels, such as DAPI and the protein of interest. In this case, the green channel, corresponding to the protein of interest, was selected. After completing the analysis, the software generated a table displaying the arithmetic mean intensity, representing the fluorescence intensity for the selected protein.
In a separate experimental set, to assess the impact of blocking TRPV4 with RN-1734) on the nuclear translocation of the transcription factor NFкBp65 in EGCs, as well as its effect on the expression of TNF-α, cells were pre-incubated with RN-1734 1 h before to exposure to TcdA or TcdB. The primary antibodies used were anti-NFкBp65 (1:500, Abcam 16502) and anti-TNF-α (1:500, Abcam 6671), along with the secondary antibody (Alexa Fluor 488, 1:400, Invitrogen). The remaining steps of the procedure were carried out as previously described, with an average of 10 images acquired per slide. The average TNF-positive fluorescence intensity was quantified using Zeiss software. The percentage of NFκB p65-positive cells was determined by counting 100 cells per coverslip across five separate experiments, focusing on the “hot areas” (regions exhibiting intense staining).
To evaluate the effect of TRPV4 activation by its agonist, GSK 1,016,790 A (50 nM/ml), and inhibition by its antagonist, RN-1734 (100 µM), on caspase-3 activation in EGCs, cells were pre-incubated with either GSK 1,016,790 A or RN-1734 for 1 h before exposure to TcdA or TcdB. The primary antibody was anti-cleaved Caspase-3 (1:400, Millipore AB3623), followed by the secondary antibody Alexa Fluor 488 (1:400, Invitrogen). The remaining steps were conducted as previously described, with an average of 10 images acquired per slide. The cleavage caspase-3-positive fluorescence intensity was quantified using Zeiss software following the procedure described above.
Realtime-glo annexin V apoptosis assayCell death was assessed using a live cell real-time assay (Realtime-Glo annexin V apoptosis assay, Promega, JA1000), following the manufacturer’s instructions. EGCs (104 cells/well), at passage 20, were seeded in white tissue culture-treated 96-well plates (Falcon, solid white bottom). These cells were incubated with TcdA, TcdB or both TcdA and TcdB simultaneously for 18 h, in either the absence or presence of RN-1734, added 1 h before exposure to the C. difficile toxins. Subsequently, 200µL of 2x detection reagent (containing 2µL of annexin NanoBit substrate, 2 µL of CaCl2, 2µL of annexin V-SmBit, and 2µL of annexin V-LgBit in 1000µL of pre-warmed, supplemented DMEM) was added to each well. The cells were then incubated at 37 °C in a humidified incubator under 5% CO2. Luminescence readings were recorded using a luminometer (NanoLuc technology ready, Promega). The intrinsic luminescence of the reagent (no-cell, no-compound background control) was subtracted from the sample luminescence signals. The adjusted values were normalized against the mean of control cells to obtain the relative luminescent units (RLUs).
Assessment of intracellular calcium levelsTo assess intracellular Ca²⁺ levels, EGCs were seeded at a density of 60,000 cells per well in black 96-well plates. The intracellular Ca²⁺ levels were measured using the Fluo-8 no wash calcium assay kit (Abcam, ab112129) following the manufacturer’s instructions. In brief, 100 µL of Fluo-8 dye-loading solution was added to each well and incubated at 37 °C for 30 min. This was followed by an additional 30-minute incubation at room temperature to ensure optimal dye uptake. Afterward, the cells were treated with TcdA and TcdB, and/or the TRPV4 antagonist, with the antagonist being added one hour prior to toxin incubation. Fluorescence intensity was monitored at an excitation/emission wavelength of 490/525 nm at different time points using the Infinite 200 PRO-Tecan plate reader. The fluorescence intensity of Fluo-8 (Ex/Em = 490/525) directly corresponds to the increase in intracellular calcium levels.
Statistical analysisAll quantitative results obtained in this research were expressed as mean ± standard error of the mean (SEM). Statistical analysis of the data was performed using GraphPad Prism software, version 8.0. The student’s t-test was used to compare the means between two groups, and the analysis of variance (ANOVA) followed by Tukey’s test was used to compare the means among three or more groups. A p-value ≤ 0.05 was considered statistically significant.
留言 (0)