Chemicals
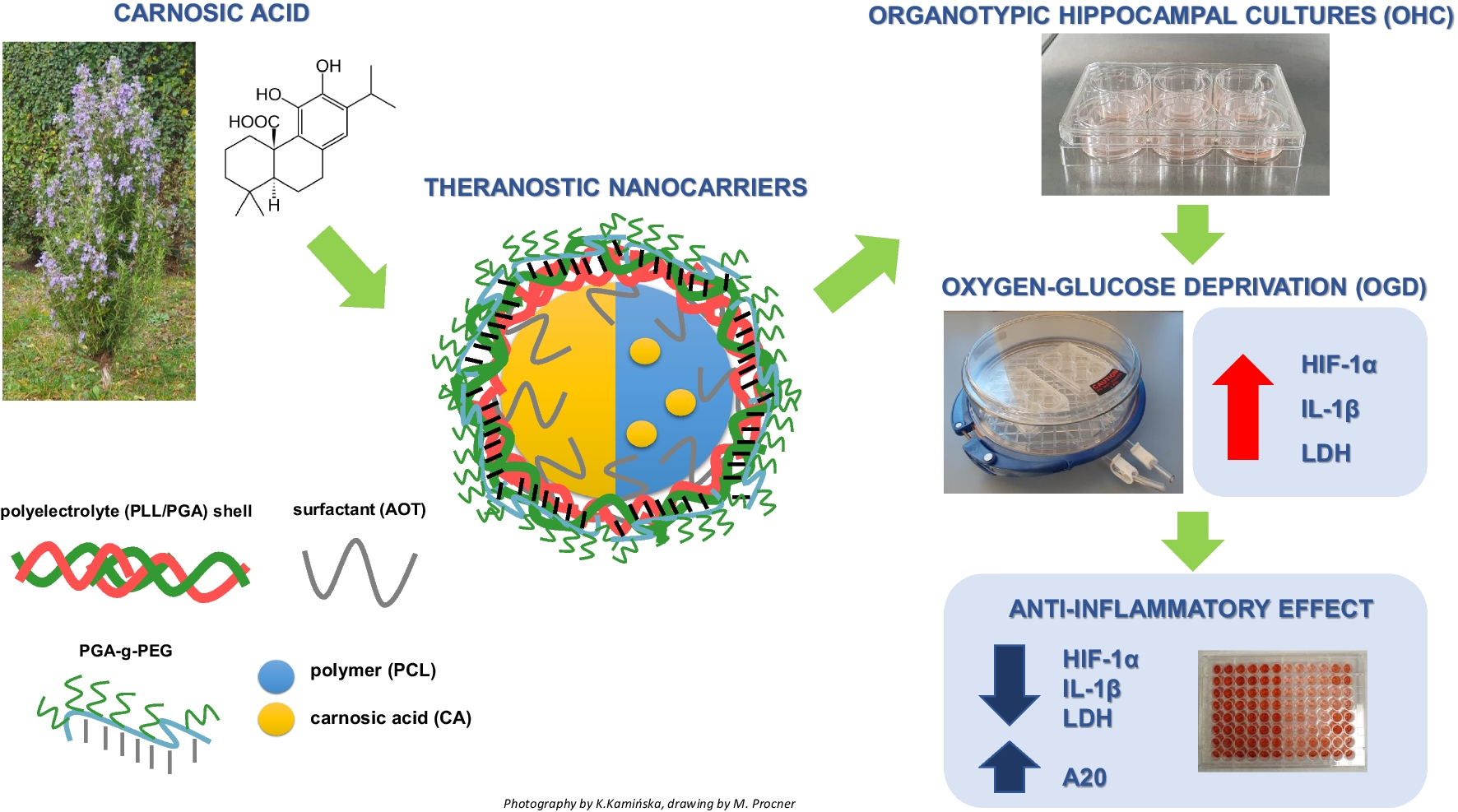
Carnosic acid was received from ChemNorm Biotech Co., Ltd. Polyelectrolytes used to prepare multilayered shells of nanocarriers were as follows: poly-l-lysine hydrobromide (PLL, Mw 15,000–30,000), poly-l-glutamic acid sodium salt (PGA, Mw 15,000–50,000) both from Sigma–Aldrich, gadolinium-labelled poly-l-lysine (PLL-Gd, Mw 22,000) from BioPAL, pegylated polyanion PGA-g-PEG (g ~ 30% and PEG, Mw 5000), and rhodamine-labeled polycation PLL-Rod were synthesized according to the protocol described previously [15, 16]. Docusate sodium salt (AOT), polycaprolactone (PCL, Mw 14,000), chloroform, and sodium chloride were obtained from Sigma-Aldrich. Ethyl alcohol (99.8%) was purchased from Avantor Performance Materials Poland S.A., while ultra-purified water was obtained using the Direct-Q5 UV purification system from Millipore (Warsaw, Poland). All chemicals and solvents were of analytical grade and used as received.
Preparation of Polyelectrolyte Multilayer Nanocarriers
Multilayered nanocarriers with two types of core (polymeric and nanoemulsion ones) were formulated by previously developed methods [17,18,19,20]. Polymeric and nanoemulsion cores loaded with carnosic acid were formed by the spontaneous-emulsification solvent evaporation (SESE) method. The oil phase based on easily evaporative organic solvent chloroform contains carnosic acid (100 mg/ml), AOT (330 mg/ml), and in the case of polymeric core PCL (10 mg/ml), while the aqueous phase contains PLL (0.2 mg/ml) and NaCl 0.09%. The nanoemulsions were formed by the addition of 0.1 ml of oil phase mixed with 10 ml of absolute ethanol to 200 ml of agues phase during continuous stirring. The polymeric or nanoemulsion cores were formed after the evaporation of the organic solvents by continuous stirring, where the final concentration of chloroform did not exceed 0.04 mg/l [15]. Such formulated cores were encapsulated into a multilayered shell via a layer-by-layer technique [2,3,4] with the following polyelectrolytes: PLL, PGA/ PLL-Gd, PLL-Rod, PGA-g-PEG.
Determination of Particle Size and Zeta Potential and Encapsulation Efficacy
The hydrodynamic size and zeta potential were determined from dynamic light scattering measurements and electrophoretic mobility, respectively, using Zetasizer Nano ZS, Malvern Panalytical Instruments, UK. All measurements were performed at 25 °C in 0.09% NaCl, and each value was received as an average of at least three subsequent measurements with 20 runs. The nanocarriers were analyzed also by a cryo-scanning electron microscope (cryo-SEM) Jeol JSM-7600F Field Emission Scanning Electron Microscope (FESEM) (Jeol Ltd., Tokyo, Japan). Confirmation of encapsulation and determination of encapsulation efficacy were based on UV–vis spectrometry measurements by UV-1800 spectrophotometer, Shimadzu Corporation, Kyoto, Japan.
Animals
Sprague–Dawley rats (226–275 g) were purchased from Charles River (Sulzfeld, Germany) and maintained under conventional laboratory conditions at a temperature of 23 °C, with a 12-h light/12-h darkness cycle (commencing at 08:00), and provided ad libitum access to water and food. Following an acclimatization period, the stage of the estrous cycle in the female rats was determined by measuring vaginal wall impedance (model MK-12UB, Ugo Basile, Gemonio, Italy). On the day of proestrus, female rats were paired with males for 12 h, and the presence of sperm in vaginal smears was assessed the subsequent morning. All experimental procedures adhered to the guidelines stipulated by the Committee for Laboratory Animal Welfare and Ethics of the Maj Institute of Pharmacology, Polish Academy of Sciences, Krakow, Poland. The protocol for generating the organotypic hippocampal cultures (OHCs) is in line with the European Union (Directive 2010/63/EU, amended by Regulation (EU) 2019.1010) guidelines on the ethical use of animals and according to national regulations. All experiments were conducted according to the principles of the Three Rs, and all efforts were undertaken to minimize the usage of animals and alleviate any potential suffering.
Organotypic Hippocampal Cultures
OHCs were established by the Stoppini et al. [21] protocol, incorporating slight modifications as described by Bryniarska-Kubiak et al. (2023) and Tylek et al. (2023) [22, 23]. The cultures were prepared from 6- to 7-day-old Sprague Dawley pups. After decapitation, the isolated brains were immediately placed into a sterile ice-cold working buffer consisting of 96% HBSS, 3.5% glucose, and 0.5% penicillin/streptomycin (all reagents obtained from Gibco, Waltham, MA, USA). The hippocampi were isolated and placed on Teflon disks, and sliced into 350-µm-thick sections using a McIlwain™ Tissue Chopper (Surrey, UK). Subsequently, selected sections were transferred into ThinCerts™ inserts with 0.4-μm pore size membranes (Greiner Bio-one, Kremsmunster, Austria) in 6-well plates containing 1 mL of initial medium enriched with 25% horse serum (50% DMEM + GlutaMax™-I, pH 7.4; 20.5% HBSS; 25% horse serum; 0.1 mg/mL glucose; 1% amphotericin B; 0.4% penicillin and streptomycin; 1% B-27 supplement; and HEPES—all reagents obtained from Gibco, London, UK). OHCs were maintained in an incubator at 37 °C under an atmosphere of 95% air and 5% CO2. The culture medium was changed every 48 h, with a gradual reduction in horse serum concentration from day 4 to day 7. By the 7th day, the medium became serum-free and was composed of 50% DMEM F-12, 44% HBSS, 0.1 mg/mL glucose, 1% B-27 supplement, 1% N2 supplement, 1% amphotericin B, 0.4% penicillin and streptomycin, and HEPES.
Oxygen–Glucose Deprivation
The OGD procedure was conducted on the 7th (OHCs) or 5th (BBB model) day after the culture establishment, following the Bryniarska-Kubiak et al. (2023) [22] protocol. OHCs or hCMEC/D3 cells were washed twice in Ringer’s solution containing 10 mM mannitol (Sigma–Aldrich, St. Louis, MO, USA). Subsequently, the membranes with OHCs or hCMEC/D3 cells were relocated to new 6- or 24-well plates respectively containing 1 mL or 300 µl Ringer’s solution and transferred into a hypoxic chamber (maintained at 37 °C; gas composition: 95% N2, 5% CO2) for 40 min. Following hypoxic exposure, the inserts with hippocampal slices or hCMEC/D3 cells were transferred back to starting serum-free plates and grown under standard conditions (37 °C; 5% CO2). Nanoparticles, either AOT, AOT-Gd, or PCL and PCL-Gd, as well as carnosic acid (CA) and nanoparticles loaded with CA were administered to the culture medium 30 min before the OGD procedure. All colorimetric or biochemical assays were conducted 24 or 48 h after OGD procedure initiation.
Propidium Iodide Cell Viability Flow Cytometry
Twenty-four hours after the OGD procedure, the hippocampal organotypic cultures were stained with propidium iodide (PI) as we previously described by Głombik et al. [24] with modifications. Briefly, hippocampal cultures were washed twice with ice-cold HBSS (Gibco, MA, USA) and transferred to 1.5-ml centrifuge tubes containing 500 μl of HBSS. After 5 min of centrifugation (1200 rpm), the cultures were incubated with 500 μl of prewarmed collagenase A solution (2 mg/ml; 30 min at 37 °C; Gibco, NY, USA), centrifuged, and incubated again with 500 μl of prewarmed trypsin (0.05%; 20 min at 37 °C; Sigma-Aldrich, MO, USA). Next, the cultures were stained with a PI solution (100 ng/ml in PBS; Sigma-Aldrich, Germany) for 15 min at room temperature. The hippocampal cells were analyzed using the BD Fluorescence-Activated Cell Sorting (FACS) Canto II System and BD FACS Diva™ v5.0.1 Software (BD Biosciences, USA) in the fluorescence channel for PE (phycoerythrin, red fluorescence). The PI-negative cells were considered to be undamaged and alive, the PI-intermediate cells were considered to be apoptotic, and the PI-positive cells were considered to be dead.
Lactate Dehydrogenase Assay
A lactate dehydrogenase (LDH) assay was conducted using a Cytotoxicity Detection Kit (Roche, Germany) as previously described [23, 25]. Briefly, 24 or 48 h after OGD, the culture medium was collected, and 50 μL of each sample was placed into a 96-well plate. Then, an equal amount of reagent mixture prepared according to the manufacturer’s instructions was mixed with the samples. After 30 min incubation at 37 °C, the intensity of the red color formed in the colorimetric assay was measured at a wavelength of 490 nm (Infinite® 200 PRO plate reader, Tecan, Zurich, Switzerland) and was proportional to the number of damaged/dead cells.
Nitric Oxide Release Assay (Nitrite Ion in Solution)
The amount of nitrite ion in solution (NO) was detected as we previously described [26], using a colorimetric Griess reaction under the protocol. An equal volume of the collected culture medium (50 μL), Griess A (0,1% N-1-naphthyl ethylenediamine dihydrochloride) and Griess B (1% sulfanilamide in 5% phosphoric acid; Sigma-Aldrich, St. Louis, MO, USA), was mixed in a 96-well plate. After 5 min incubation at room temperature, the intensity of the formed color was measured at a wavelength of 540 nm (Infinite® 200 PRO plate reader, Tecan, Zurich, Switzerland).
Enzyme-Linked Immunosorbent Assay (ELISA)
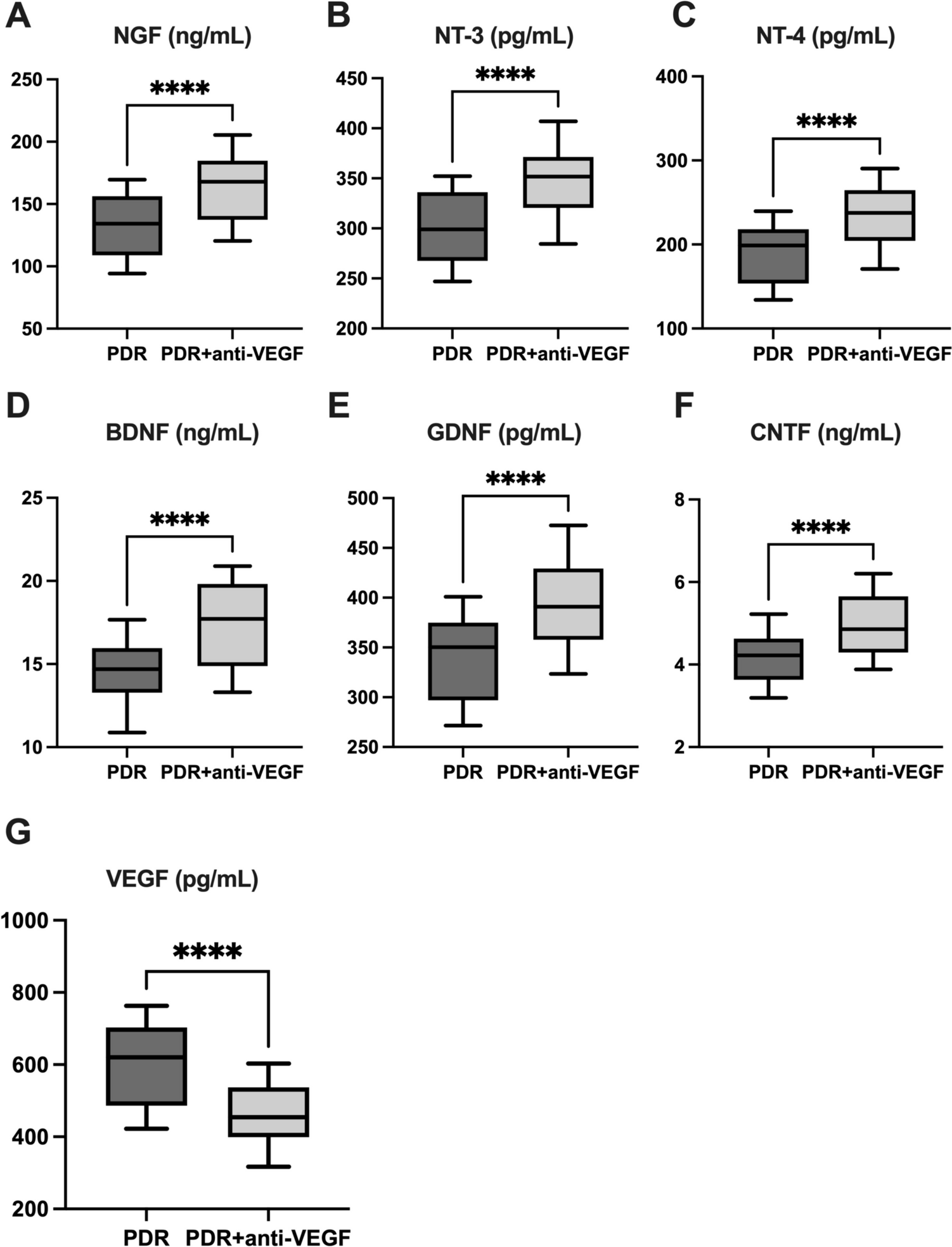
For ELISA assays, the OHC supernatants were collected 24 h or 48 h after the OGD procedure. Moreover, OHCs were lysed using 160 μL of RIPA buffer with Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, MA, USA). Protein isolation was performed, and the total concentration of the protein was assessed using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Optical density was measured at a wavelength of 562 nm using an Infinite® 200 PRO plate reader (Tecan, Zurich, Switzerland). The levels of interleukin 1β (IL-1β; obtained from Wuhan Fine Biotech Co., Ltd. Wuhan, China), interleukin 6 (IL-6; obtained from Wuhan Fine Biotech Co., Ltd. Wuhan, China), interleukin 4 (IL-4; obtained from Wuhan Fine Biotech Co., Ltd. Wuhan, China), interleukin 10 (IL-10; obtained from Wuhan Fine Biotech Co., Ltd. Wuhan, China), C–C motif chemokine ligand 2 (CCL2; obtained from Wuhan Fine Biotech Co., Ltd. Wuhan, China), C–C motif chemokine ligand 3 (CCL3; obtained from Wuhan Fine Biotech Co., Ltd. Wuhan, China), C–C motif chemokine ligand 5 (CCL5; obtained from Wuhan Fine Biotech Co., Ltd. Wuhan, China), and C-X-C motif chemokine ligand 10 (CXCL10; obtained from Wuhan Fine Biotech Co., Ltd. Wuhan, China) were measured in the collected supernatants. The hypoxia-inducible factor 1α (HIF-1α; obtained from Wuhan Fine Biotech Co., Ltd. Wuhan, China), NFκB p65 Total/Phospho (NFκB p65 Total/Phospho; obtained from Thermo Fisher Scientific, CA, USA), and A20 (Rat Tumor Necrosis Factor Alpha Induced Protein 3(TNFaIP3) or A20 obtained from Wuhan Xinqidi Biological Technology, China) were assessed in the cell lysates isolated from OHCs. All tests were performed using commercially available enzyme-linked immunosorbent assays (ELISA) in accordance with the manufacturer’s protocols. The detection limits were as follows: IL-1β < 18.75 pg/mL, IL-6 < 37.5 pg/mL, IL-4 < 9.375 pg/mL, IL-10 < 18.75 pg/mL, CCL2 < 9.375 pg/mL, CCL3 < 18.75 pg/mL, CCL5 < 9.375 pg/mL, CXCL10 < 18.75 pg/mL, HIF-1α < 4.688 pg/mL, A20 < 10 ng/mL. The inter-assay precision of all ELISA kits was CV% < 12%. The intra-assay precision of all ELISA kits was CV% < 8%. The NFκB p65 Total/Phospho level was measured qualitatively as the relative concentration and normalized to the total protein concentration.
Materials for Cell Culture
Endothelial cell basal medium (EBM-2) was provided by Lonza Group Ltd. (Basel, Switzerland). Cultrex Rat Collagen I was from R&D Systems (Minneapolis, MN, USA). FluoroBrite DMEM, heat-inactivated fetal bovine serum (FBS), DPBS, penicilin-streptomycin mixture (10,000 U/mL), Trypsin–EDTA solution, HEPES, chemically defined lipid concentrate and human basic fibroblast growth factor (bFGF) were supplied by Gibco (Invitrogen, Paisley, UK). Sulforhodamine B acid chloride and ascorbic acid were purchased from Sigma-Aldrich Chemie GmbH (Germany). Collagen Coating Transwell Inserts (pore size 1 µm) and suitable 24-well plates were obtained from Corning Life Sciences (New York, USA). All materials were used without further purification.
hCMEC/D3 Cell Culture
Human cerebral microvascular endothelial (hCMEC/D3) cell line (obtained from Sigma-Aldrich Chemie GmbH, Germany), as an in vitro model of BBB was prepared following the procedure described by Łukasiewicz et al. [27]. hCMEC/D3 cells were maintained in 75 cm2 flasks pre-coated with 150 µg/ml rat collagen type I (at least 1 h before use and placed in the incubator) in endothelial basal medium-2 (EBM-2) supplemented with 5% (v/v) heat-inactivated FBS, 1% (v/v) penicillin–streptomycin mixture, 1% (v/v) HEPES, 1% (v/v) chemically defined lipid concentrate, 5 µg/ml ascorbic acid, 500 ng/ml hydrocortisone, and 1 ng/ml bFGF (prepared according to manufacturer’s protocol). Cells were cultured at 37 °C under an atmosphere of 5% CO2. The culture medium was replaced with fresh medium every 2 days. After reaching 80% confluency, hCMEC/D3 cells were passaged using 0.25% Trypsin/EDTA solution (37 °C, 15–20 min) and centrifugated (900 rpm, 8 min). Next, the pellet was resuspended in fresh EBM-2 medium, counted using a Bürker chamber, and seeded in flasks or plates in suitable density for further experiments. Cells were used for experiments between passages 27 and 35.
hCMEC/D3 Cell Treatment
hCMEC/D3 cells were seeded at a density of 4.5 × 104 cells/well on Transwell inserts precoated with rat collagen type I (150 µg/ml, 1 h before use in 37 °C) in 24-well plates (Corning, USA) and were cultured for 5 days at 37 °C, 5% CO2, and 95% humidity to form confluent monolayer. The culture medium was changed every 2 days and before the treatment. The experiments were performed 5 days after seeding the cells, allowing sufficient time to develop the junctions between cells [27]. After the OGD procedure, the apical chambers (Transwell inserts) were filled with 75 µl or 150 µl of rhodamine-labeled nanoparticles (AOT-ROD/CA and PCL-ROD/CA) and supplemented with EBM-2 medium up to 750 µl. The well under the insert consisted of 750 µl fresh Fluorobrite DMEM, which ensures undisturbed fluorescence measurements. Additionally, rhodamine B alone (diluted in ethanol:water 50% mixture) and 0.015 M NaCl were placed in selected inserts as a control. Plates were incubated at 37 ℃ under standard conditions. At different time points, 50 µl of medium from wells was collected in 96-well black plates and additionally replenished with 50 µl of Fluorobrite medium. Likewise, the content of wells under the inserts was refilled with fresh Fluorobrite medium every time. The penetration ability was determined by measuring fluorescence intensity at the excitation and emission wavelengths of 558 nm and 586 nm, respectively, using an Infinite® 200 PRO plate reader (Tecan, Zurich, Switzerland). All measured fluorescence values were corrected by subtracting the background signal from the pure Fluorobrite medium.
TEER Measurements
Trans epithelial electrical resistance (TEER, in Ω·cm2) was measured every day to confirm the presence of tight monolayer of hCMEC/D3 cells using Millicell ERS-2 Electrical Resistance System (Epithelial Volt–Ohm Meter, Merck Millipore, Burlington, MA, USA). Before the measurements, the electrode was sterilized in 70% ethanol for 15 min. Wells with collagen-coated Transwell inserts without cells were considered a control in TEER measurements. The resistance was calculated by multiplying the Transwell insert’s area with the obtained resistance value. The model of the blood–brain barrier is completely prepared for further studies when the resistance reaches the highest value among all measurements.
Statistical Analysis
Data were analyzed using the Statistica 14 software (StatSoft Inc., Tulsa, OK, USA). The analysis of variance (one-way ANOVA) and post hoc Duncan’s test for multiple comparisons were used to show statistical significance with assumed p < 0.05. The results were obtained from independent experiments carried out under the same conditions and are presented as the mean ± SEM. All graphs were prepared using GraphPad Prism 9.

留言 (0)