
記住我


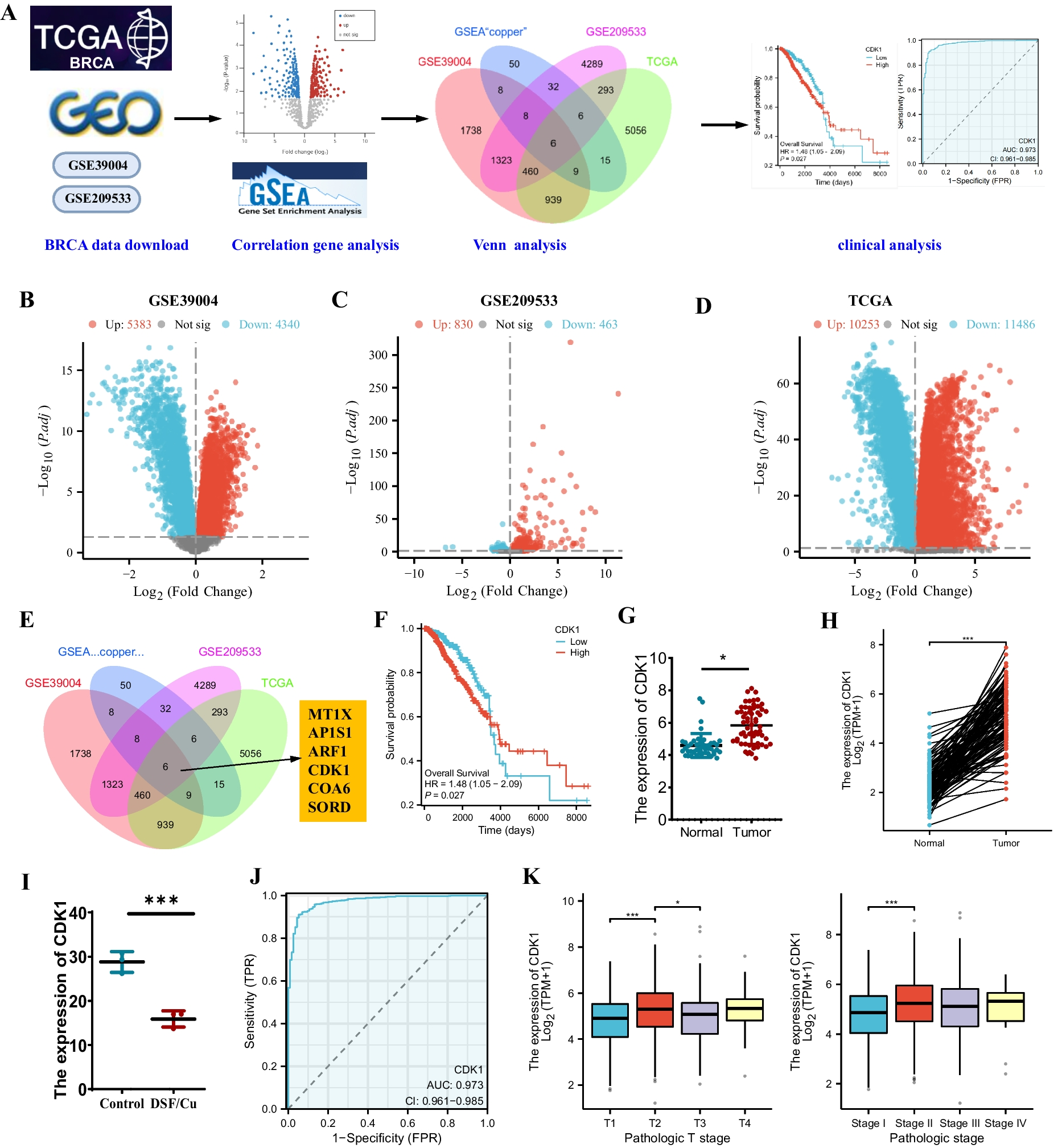


The Cancer Genome Atlas (TCGA) project provided the RNA sequencing data and clinicopathological data concerning BRCA (TCGA-BRCA). Enclosed in the datasets were 113 samples from normal tissues adjacent to the tumors, as well as 1109 tumor samples. Details concerning 1095 subjects was obtainable, comprising futime, fustat, individual's age, sex, cancer grade, stage, T stage, M stage, and N stage. Additionally, the BRCA transcriptome sequencing dataset GSE39004 was retrieved from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/), which included 61 primary BRCA tissue samples. The GSE39004 dataset was integrated with the TCGA-THCA dataset, and the "combat" algorithm was employed to remove batch effects (Fig. S1) (Wang et al. 2022a, b).
Single-cell sequencing data download and preprocessingThe single-cell sequencing dataset GSE180286 specific to BRCA was sourced from the GEO database. Five primary BRCA tumors (GSM5457199, GSM5457202, GSM5457205, GSM5457208, GSM5457211) were selected for analysis, and processing of the data was achieved with the R package "Seurat". Criteria for quality assurance involved ensuring nFeature_RNA was limited between 200 and 5000, in addition to maintaining the mtRNA percentage below 20%, and a compilation of the top 2000 genes displaying significant variability was established through variance analysis (Meng et al. 2021).
Gene ontology (GO) and kyoto encyclopedia of genes and genomes (KEGG) analysisAnalysis of GO and KEGG pathway functions in the CAF cell lineage was accomplished through the utilization of the cluster Profiler R package (version 3.14.3), focusing on enriching biological processes (BPs), molecular functions (MFs), cellular components (CCs), and identified marker gene-enriched pathways. A significance threshold of less than 0.05 was deemed as statistically important (Yu et al. 2012).
Genetic mutation analysisInformation regarding genetic mutations, encompassing somatic changes and alterations in copy numbers (CNV) within the BRCA gene, was acquired from the GDC TCGA-BRCA database. The examination and visualization of somatic mutations in TC were executed by employing the maftools R package (version 2.6.05). Moreover, human chromosomal CNV profiles of CRGs were graphically represented through the RCircos R package (1.2.2 version). It is recommended to break down long sentences into shorter ones to enhance readability (Li et al. 2022).
Formation of protein–protein interaction (PPI) networksThe establishment of the PPI network employed the resources from the STRING database to investigate the potential interactions among the proteins encoded by the 30 identified CAF feature genes. Setting the interaction threshold at 0.4 corresponded to a medium level of confidence, and both known and predicted protein–protein associations were included. To visualize and analyze the PPI network, we employed Cytoscape software, which allowed for an in-depth exploration of the co-expression and interaction relationships between the proteins involved in extracellular matrix organization and tumor invasion pathways.
Correlation analysisThe application of the ggcorrplot software (v0.1.3) aided in the computation and depiction of the potential correlation between characteristic genes of CAF and gene expression present in BRCA for improvement (Li et al. 2022).
Consensual clustering and genetic clusteringPatients were assigned to specific molecular subtypes by utilizing the consensus clustering approach after the selection of characteristic genes related to CAF. The software "ConsensusClusterPlus" was applied for identifying the stability and quantity of clusters, with 1000 repetitions conducted for stability assurance. A catalogue of DEGs from consensus clustering was disclosed by applying the limma R software package and conditions of |log2(fold change)|> 1 and false discovery rate (FDR) < 0.05. Subsequently, patients were classified into diverse gene subgroups (Class A, Class B, and Class C) through unsupervised clustering, driven by the expression patterns of prognostic DEGs, to enable subsequent analyses. For a more thorough investigation into the clinical implications of consensus clustering and gene clustering techniques, the correlation between molecular subgroups, clinicopathological features, and prognostic outcomes was examined (Wilkerson and Hayes 2010). Clinical features included age, gender, TNM staging, and grading. Additionally, execution of the survival analysis using the Kaplan–Meier (KM) method was accomplished utilizing the survival toolkit available in the R software environment.
Gene set variation analysis (GSVA)GSVA is a popular approach for estimating modifications in pathway and BP activity in a dataset of gene expression samples. This method employs the "GSVA" R package to interpret BP differences between the two CAF scoring subtypes. The gene set denoted as "c2.cp.kegg.v7.4.symbols.gmt" was sourced from the MSigDB database for the purpose of GSVA scrutiny (Hänzelmann et al. 2013).
Relationship between molecular subtypes and TMEValidation of TME attributes within diverse molecular categories involved the utilization of marker gene sets (C2.CP.KEGG (186 gene sets) and C5.GO. gene Ontology (10561 gene sets)) extracted from the MSigDB repository for GSVA analysis. An observed statistical significance corresponded to adjusted p-values under 0.05. The application of the deconvolution algorithm (CIBERSORT) aided in the computation of immune cell infiltration amounts (TICs) within individual BRCA specimens, utilizing the gene expression attribute matrix from the CIBERSORT platform. Comparing the gene expression level matrices of TCGA-THCA and GSE27155 datasets with the feature matrix of TICs led to the formulation of a TICs proportion matrix in BRCA tissues. CIBERSORT p-values < 0.05 were deemed appropriate for additional investigation. Additionally, the methodology used for determining the TICs in individual BRCA specimens involved the application of the single-sample gene set enrichment analysis (ssGSEA) algorithm (Wang et al. 2022a, b).
Construction of CAF risk scoring modelA univariate COX regression analysis was executed on the DEGs from the molecular subtypes of CAFs to pinpoint genes related to the survival of individuals diagnosed with BRCA. Subsequently, using consensus clustering analysis based on the expression of prognostic DEGs, individuals were categorized into different CAF gene subtypes, including the A subtype, the B subtype, and the C subtype. Next, employing the "caret" package in R, we arbitrarily divided every BRCA case from TCGA-BRCA and GSE39004 databases into training set (n = 539) and test set (n = 538) at a 1:1 ratio. The CAF Risk scoring system was developed in the training dataset by applying LASSO COX regression analysis through the "glmnet" package in R to reduce overfitting risks. The trajectory of every separate factor was examined and cross-validated for model establishment. A prognostic CAF Risk scoring system was formulated by applying Multivariate Cox analysis to pinpoint potential risk genes in the training set. Below is the formula for assessing CAF risk score: CRGs risk score = ∑ (Exp_i * coeff_i), where Exp_i represents the expression of key CAF-related risk genes, and coeff_i represents the risk coefficient. Examination of correlations was employed to assess the interrelation between risk scoring and diverse molecular or gene subtypes. Categorized by the median risk score, the group of 1077 BRCA patients was split into a high-risk group (n = 522) and a low-risk group (n = 555). Subsequently, utilization of KM survival analysis aimed to evaluate the variance in survival rates among the high-risk and low-risk cohorts. Finally, survival analysis and the creation of ROC curves were facilitated by classifying the test set and combined set into high-risk and low-risk divisions (Wang et al. 2022a, b).
Correlation of CAF risk assessment model with TMEIn R language, boxplots were employed for evaluating the CAF expression in both high-risk and low-risk score categories, determining the TICs abundance within the TME of each C specimen through CIBERSORT, followed by correlation analysis to explore the link between TICs and prognostic risk genes (Sha et al. 2022).
Construction of column line plots and validationUtilizing survival analysis methods in R packages, the independence of the CAF model from clinical features was evaluated through univariate and multivariate Cox regression analyses. Subsequently, relying on the multivariate Cox regression coefficients related to CAF attributes and clinical factors within the TCGA training group, we constructed column line plots and computed the concordance index (C-index) to verify the forecast capability of these graphs. For the validation process, calibration curves were generated by bootstrapping with 1000 iterations to evaluate the concordance between anticipated 1-year, 3-year, and 5-year overall survival (OS) probabilities and the actual observed values (Zheng et al. 2021).
Clinical sample collectionThis study collected tissue samples from 55 BRCA patients, including tumor tissue and normal breast tissue located a minimum of 5 cm from the tumor. The clinical attributes of 55 individuals with BRCA are outlined in Table S1. All study participants agreed to the publication of the research results. Patients who had received cancer-related treatments such as radiation therapy, chemotherapy, and hormone therapy were excluded from the study. Patients with major chronic diseases, recent major surgeries or medical interventions in pregnancy or lactation, as well as those with immune disorders, were also excluded. These criteria ensured data consistency and accuracy of the research results. This study was granted permission by the the Clinical Ethics Committee of The First Hospital of China Medical University. Official authorization for animal experimentation protocols was granted by the Animal Ethics Committee of The First Hospital of China Medical University (No. CMUXN2022131) (Primac et al. 2019).
Isolation of NF (normal fibroblasts) and CAFIn simple terms, finely mincing and enzymatically digesting both tumor and non-tumor samples by employing type I collagenase (17100017, Gibco, USA) is essential prior to their cultivation in DMEM medium (11965092, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, 10100, GIBCO, USA). The ideal method for cell growth involves maintaining them in a humid atmosphere at 37°C, with 5% CO2 supplementation until NF/CAF attaches to the culture dish, and primary NF/CAF should be used before passage 6. Subsequently, immunofluorescence should be used to observe the expression of fibronectin as a fibroblast marker and cytokeratin as an epithelial cell marker to identify the isolated primary NF/CAF (Wen et al. 2019).
Cell cultureWuhan Pronas Life Technologies Co., Ltd. (CL-0150A, China) supplied the human BRCA cell line MDA-MB-231. Cell cultivation took place in the RPMI 1640 medium (11875119, GIBCO, USA) encompassing 10% FBS (10100, GIBCO, USA) and 1% penicillin–streptomycin (10378016, GIBCO, USA). Incubation of cells occurred in a humidified environment with 5% CO2 at a temperature of 37 degrees Celsius. When undertaking lentiviral cell transfection, a 6-well plate was populated with 5 × 105 CAF cells. At the point of cell density hitting 70–90%, the cells were exposed to a medium comprising an optimal dosage of lentivirus (MOI = 10, working strength around 5 × 106 TU/mL) and 5 μg/mL polybrene (sourced from Merck, TR-1003, USA). Post 4 h of transfection, an identical medium amount was included to lessen the polybrene concentration, followed by a switch to fresh medium after 24 h. Fluorescent luciferase was used to assess gene transfection and cells with stable transfection were selected employing 60 μg/mL puromycin (Sangon Biotech, A100339, Shanghai, China) after 48 h. Sangon Biotech (Shanghai, China) offered the lentivirus packaging assistance. The following sequences are utilized in lentivirus-mediated gene silencing: sh-NC: 5'-CCTAAGGTTAAGTCGCCCTCG-3'; sh-SDC1: 5'-CCGACTGCTTTGGACCTAAAT-3'; sh-RUNX1: 5'-CCTACGATCAGTCCTACCAAT-3' (Sohn et al. 2018).
Dual-luciferase assayThe JASPAR database (https://jaspar.elixir.no/) was utilized to predict the presence of binding sites for RUNX1 and SDC1 at their respective promoters. The effect of RUNX1 on the transcriptional activity of the SDC1 promoter was investigated by co-transfecting CAF cells with Lipofectamine 2000 transfection reagent (Catalog number: 11668019, ThermoFisher, USA) and introducing oe-NC, oe-RUNX1, sh-NC, sh-RUNX1 plasmids, along with dual-luciferase reporter gene vectors containing the SDC1 promoter sequence (AATTGTTGTAA) and their corresponding mutant binding sites (TTAACAACATT). Renilla luciferase functioned as an internal control. Subsequent to transfection for 48 h, cellular collection and lysis were performed, and the assessment of luciferase activity was carried out using the Dual-Luciferase Reporter Gene Analysis System (Promega, Madison, WI, USA). Analyzing the ratio of firefly luciferase luminescence units (RLU) to Renilla luciferase luminescence units (RLU) allowed for the assessment of the activation level of the specific reporter gene (Taniue et al. 2016).
ChIP assayThe enrichment status of RUNX1 within the SDC1 gene promoter region was examined using a ChIP kit (Catalog number: KT101-02, Saicheng Biotechnology Co., Ltd., Guangzhou, China). The steps involved the fixation of cells at 70–80% confluence with 1% formaldehyde at room temperature (RT) for 10 min to create cross-links between DNA and proteins, followed by sonication to shear the cross-linked DNA–protein complexes into suitable-sized fragments. Centrifugation at 13000 rpm at 4 °C was performed to collect the supernatant, which was then divided into two tubes. In one test tube, overnight incubation occurred at 4 °C with Rabbit IgG (ab172730, 1:100 dilution, Abcam, UK) as the negative control antibody, while the other tube with a specific antibody against the target protein, Rabbit anti-RUNX1(1:100, ab272456, Abcam, UK). The separation of endogenous DNA–protein complexes was accomplished using the Protein Agarose/Sepharose precipitation method, followed by reversing the cross-links overnight at 65°C. Subsequently, DNA fragments were purified through phenol/chloroform extraction for the qPCR analysis of SDC1 gene promoter segments with the primers: Forward 5'-CCACAGAAAAACGCTGCGAA-3'; Reverse: 5'-CCAGATTCTCCCGTACGCTC-3' (Nelson et al. 2006).
Cell immunofluorescence stainingAfter counting the NF/CAF cells, they were dispersed and cultured in immunofluorescence chambers. Each well contained 2 × 105 cells. Upon reaching approximately 90% cell confluence, cells were rinsed three times with ice-cold PBS. 4% paraformaldehyde was used for cell fixation, adding 1 mL to individual wells, and then incubating at RT for 15 min. Upon three rounds of PBS rinses, cells were subjected to blocking utilizing 5% BSA, followed by a 30-min incubation period. Rabbit anti-α-SMA (#19245S, 1:200, Cell Signaling Technology, USA), Mouse anti-SDC1 (ab181789, 1:500, Abcam, UK), fibronectin (ab2413, 1:250, Abcam, UK), Cytokeratin (ab53280, 1:250, Abcam, UK) primary antibodies underwent an overnight incubation at 4°C, succeeded by three rounds of PBS rinsing. Subsequently, secondary antibodies, Goat anti-rabbit IgG H&L (Alexa Fluor 488) (1:200, ab150077, Abcam, UK) and Goat anti-mouse IgG H&L (Alexa Fluor 647) (1:200, ab150115, Abcam, UK) were applied and maintained for an hour at RT, then subjected to three PBS rinses. Cellular nuclei were labeled using PI (P1304MP, Invitrogen, USA) or DAPI (D9542, Sigma) dyes in low light conditions for a duration of 15 min. Ultimately, the slides were arranged with a fluorescence quencher and visually examined and imaged with a fluorescence microscope by Olympus, a Japanese manufacturer. The quantitative analysis was executed with Image-Pro Plus 6.0 software after three washes with PBS under dark conditions (Beneit et al. 2016).
Western blotUtilization of RIPA lysis buffer (P0013B, Beyotime, Shanghai) encompassing 1% PMSF enabled the lysis of cells to obtain total proteins. Cell membrane proteins were extracted using the ProteoPrep® Membrane Extraction Kit (PROTMEM-1KT, MERCK). Each sample's protein concentration was gauged through the utilization of the BCA assay kit (P0011, Beyotime, Shanghai). The SDS-PAGE gels were developed within the 8% to 12% concentration range, adjusting the molecular weight of the identified protein bands. Matching quantities of protein samples were evenly inserted into all lanes using a micropipette to perform the process of electrophoresis separation. Transferring of proteins from the gel was done onto a PVDF membrane (1620177, BIO-RAD, USA), followed by blocking the membrane with 5% skim milk at RT for 1 h. Primary antibodies against α-SMA (#19245S, Cell Signaling Technology, USA), FAP-α (ab207178, Abcam), SNAI1 (PA5-23482, Invitrogen), MMP2 (ab86607, Abcam), SDC1 (ab128936, Abcam), RUNX1 (ab240639, Abcam), MMP9 (ab76003, Abcam), and GAPDH (ab8245, Abcam) were incorporated, followed by an overnight incubation of the membrane at a temperature of 4°C. Three 5-min washes were carried out on the membrane with 1 × TBST at RT. HRP-conjugated secondary antibodies, goat anti-rabbit IgG (ab6721, 1:2000) or goat anti-mouse IgG (ab6728, 1:2000), were included and kept at RT for 1 h. These antibodies were procured from Abcam in the UK, Cell Signaling Technology in the USA, and Invitrogen in the USA. Three wash cycles were performed on the membrane using 1 × TBST buffer at RT, lasting 5 min per cycle. Deployment of the ECL substrate (1705062, Bio-Rad, USA) resulted in the detection of protein bands, which were then imaged using the Image Quant LAS 4000C Gel Imaging System (GE, USA). The experiment incorporated GAPDH as an internal reference, and the relative expression quantity of the protein was determined by comparing the intensity of the target band with that of the reference band, enabling the evaluation of different protein expression levels (Wu and Yi 2018). The trial was reiterated three times for accuracy assessment.
ELISASupernatants from NF/CAF groups were collected and subjected to a human SDC1 ELISA assay kit (ab46506) for detection. This experiment aimed to detect the expression of secreted protein SDC1 following strict operational procedures (Fang et al. 2021).
Collection of NF/CAF conditioned mediumSeeded in 100 mm culture dishes, CAF and NF cells were cultured for 24 h at a cell density of 1 × 106 cells/mL, and the medium for culture was retrieved post a PBS rinse of the cells. Every well was supplied with 8 mL of serum-free medium, and post a 2-day incubation, the conditioned medium was amassed and strained using a 0.2 μm syringe filter to eradicate any remaining cells and detritus. To neutralize SDC1 in the conditioned medium of CAF, 25 μg/mL of human SDC1 antibody (ab128936) or its immunoglobulin G (IgG, ab109489) were introduced into the conditioned medium. Pursuing a 1-h incubation at RT, the medium was applied to the BRCA cells (Sun et al. 2021).
Cell scratch assayThe addition of 5 × 105 cells per well was followed by a 24-h culture period. In the next step, the medium for cultural growth was disposed of, and with the aid of a sterile pipette tip, a level scratch was meticulously crafted at the rear of the well, subsequently swapped with serum-exempt culture medium. Distances of the wounds were inspected utilizing an optical microscope (Leica, DM500) after 0 and 48 h of culture. Images were captured under an inverted microscope. The analysis of the scratch width in each well was performed using Image J software, and the cell migration capability was assessed through the comparison of scratch widths across the groups. The actual cell migration distance was determined by assessing the relative distance of cell migration to the scratch area measured from the original cell scratch area. Three rounds of experimentation were executed (Wei et al. 2020).
Transwell assayEvaluation of cell invasion potential was conducted via the Transwell examination. First, ECM gel (EHS matrix E1270-1ML, Sigma) was refrigerated at 4°C overnight and subsequently thinned in medium devoid of serum at a ratio of 1:9 to achieve a concentration of 1mg/ml. Subsequently, each 24-well Transwell chamber (354480, Shanghai Yuhui Biotechnology Co., Ltd., China) was treated with 40 μl of ECM gel on the polycarbonate membrane and incubated at 37°C with 5% CO2 for 5 h to promote gel polymerization. After the gel had polymerized and formed, excess liquid was removed, and 70 μl of pure DMEM medium was introduced into individual chamber. The matrix gel was rehydrated by undergoing a 0.5-h incubation at 37°C in a humid incubation chamber as the subsequent step. The surplus culture medium was removed, and a 24-h serum-starvation period was applied to the cells. Upon completion of centrifugation, the cells were retrieved and reconstituted in DMEM medium minus FBS to achieve a concentration of 2.5 × 105/ml. Subsequently, the hydrated basement membrane of the upper chamber welcomed 0.2ml of cell suspension and pre-cooled DMEM medium with a 10% FBS concentration was introduced into the lower chamber, totaling 700 µl. Upon completion of a 24-h incubation interval under 37°C with 5% CO2 saturation, the compartment was eliminated, and cells present on the upper membrane and basal membrane were wiped away using a wet cotton swab. Subsequent to the fixation process utilizing methanol lasting 30 min, the cells underwent staining with 0.1% crystal violet dye for a period of 20 min. Following air-drying, observation using an inverted microscope and capturing of images occurred. The experiment was performed three times, and proportions of migrating cells through the membrane were calculated by selecting five arbitrary fields randomly (Li et al. 2020).
Nude mouse tumor modelChosen were 42 female BALB/c nude rodents, 6 weeks of age (401, Beijing Vital River Laboratory Animal Technology Co., Ltd.), and housed under controlled conditions in an SPF-grade animal facility, maintained at 60–65% humidity and temperatures ranging between 22–25°C, following a 12-h cycle of light and darkness, with ad libitum access to sustenance and water. Post a week of acclimatization diet, the animal ethics commission sanctioned the experimental procedures and guidelines for animal utilization.
The animal models were separated into the following classifications: BC + NF group (injection of MDA-MB-231 and NF mixed solution); BC + CAF group (injection of MDA-MB-231 and CAF mixed solution); BC + CAF-sh-NC group (injection of MDA-MB-231 and CAF mixed solution infected with sh-NC lentivirus); BC + CAF-sh-SDC1 group (injection of MDA-MB-231 and CAF mixed solution infected with sh-SDC1 lentivirus); BC + CAF-oe-NC group (injection of MDA-MB-231 and CAF mixed solution infected with oe-NC lentivirus); BC + CAF-oe-SDC1 group (injection of MDA-MB-231 and CAF mixed solution infected with oe-SDC1 lentivirus). Each group consisted of 6 nude mice. On the 45th day after xenograft transplantation, the experimental animals were euthanized, and tumors and mouse lungs were collected for further study.
Subcutaneous Xenograft Tumor Model: MDA-MB-231 cells (1 × 106) were combined with an equivalent quantity of NF or CAF in 200μl PBS: Female nude mice, 4 weeks old, received subcutaneous injections of Matrigel prepared at a ratio of 1:1. Tumor volume (V = length × width2 × 0.5) was monitored weekly.
Lung Metastasis Model: MDA-MB-231-luciferase (1 × 106 cells) were blended with NF or CAF cells in 200μl of PBS at an equal ratio: Administered via the tail vein of nude mice, the 1:1 Matrigel mixture was coupled with a 0.2 mL cell suspension. After 4 weeks, the nude mice were placed in an IVIS Lumina XR imaging chamber (PerkinElmer, Waltham, MA, USA) for white light and bioluminescence imaging to observe lung metastasis. Subsequently, cervical dislocation was utilized for the euthanization of the mice, leading to the retrieval of their lungs. The calculation of the mean liver metastases count was executed by conducting H&E staining (Zhang et al. 2014; Vanden Borre et al. 2014).
H&E stainingLung samples were obtained from nude mice carrying tumors and then preserved using 10% neutral formalin. Subsequently, the fixed lung tissues underwent embedding in paraffin prior to sectioning. These sections were deparaffinized with xylene and stained with hematoxylin, followed by eosin staining. After being rinsed with deionized water, a graded series of ethanol was used as a dehydrating agent and cleared with xylene. Neutral resin fixation was applied to the air-dried sections for subsequent examination under an optical microscope (Wei et al. 2019).
Immunohistochemical stainingTumor tissues from nude mice, BRCA tissues, and corresponding normal adjacent tissues were procured. Formalin fixation was employed on the specimens, leading to the creation of paraffin sections with a thickness of 4 μm. Afterward, the samples were deparaffinized to water and subjected to standard immunohistochemical staining procedures. The antibodies used included α-SMA (#19245S, 1:250, Cell Signaling Technology, USA), SDC1 (Abcam, ab128936, 1:500), SNAI1 (PA5-23482, 1:200, Invitrogen), MMP2 (Abcam, ab86607, 1:200), and MMP9 (Abcam, ab76003, 1:500), all purchased from Abcam and Invitrogen. The assessment of staining outcomes involved the random selection of 5 regions through the microscope, with the quantification of cells displaying positive staining. Every assay was executed threefold (Wen et al. 2019).
Statistical applications and methodologies for data analysisThe analysis and manipulation of data were executed utilizing R 4.2.1, in conjunction with RStudio version 4.2.1 as the integrated development environment. File processing was executed employing Perl 5.30.0. The analysis of networks was executed with the assistance of Cytoscape version 3.7.2. Descriptive statistics were displayed as Mean ± Standard Deviation, with unpaired Student's t-test or Wilcoxon test utilized to compare normally distributed data across two groups, whereas one-way ANOVA followed by Tukey's post-hoc analysis was applied for comparisons among several groups. A P-value < 0.05 indicated statistical significance.
A logarithmic transformation was applied to meet the normality assumption for non-normally distributed data. Multiple imputation methods were primarily used to handle missing data to maintain data integrity and minimize bias. Additionally, to manage the surge of type I errors in multiple contrasts, the FDR control method was utilized to reduce the likelihood of chance findings, ensuring the rigor of statistical analysis and the reliability of results (Morris et al. 2017).
留言 (0)