
記住我

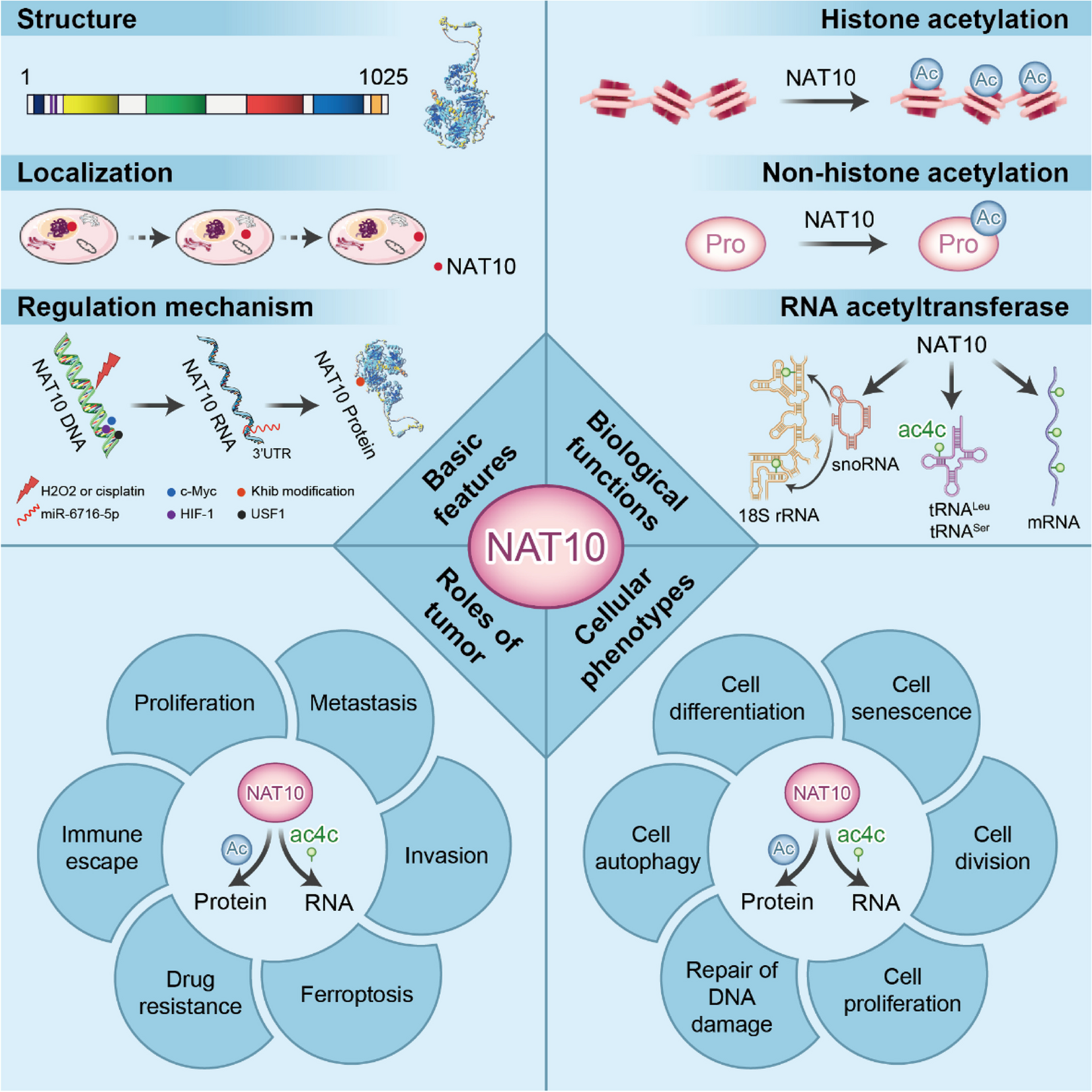


NAT10 exerts diverse roles on cellular phenotypes such as DNA damage, cell proliferation, cell senescence, cell autophagy, cell differentiation, and tumorigenesis and development. These effects may rely on either the protein acetyltransferase activity or the RNA acetyltransferase activity of NAT10 (Wang et al. 2019; Fei et al. 2021). Currently, most studies use gene overexpression (Sas-Chen et al. 2020), gene knockdown (Wei et al. 2023), or ac4C inhibitor Remodelin (Liu et al. 2020) to investigate the molecular function of NAT10. However, neither gene overexpression nor gene knockout can precisely distinguish whether NAT10 induces cellular phenotypic changes through protein acetylation or RNA acetylation. Mutation of the enzyme active functional sites is a good solution to the above problems. Sites such as K31 (Wang et al. 2019), K120 (Liu et al. 2016), K228 (Passananti et al. 2007), and K949 (Zhang and Li 2019) of NAT10 are the functional sites that mediate protein acetylation, but the enzyme active functional sites that mediate RNA acetylation by NAT10 have not been reported yet. This part summarizes the ways in which NAT10 affects different cell phenotypes as a protein acetyltransferase and an RNA acetyltransferase respectively.
NAT10 influences cellular phenotypes as protein acetyltransferaseNAT10 participates in cell division and senescence as protein acetyltransferaseNAT10 regulates the physiological activities of the centrosome through its protein acetylation activity at various stages of mitosis, thereby participating in cell division. During mitosis, the acetylation of the centrosomal protein CCDC84 at K31 is dynamically regulated by both SIRT1 deacetylase and NAT10. From G1 to mid-mitosis, SIRT1 mediates the deacetylation of CCDC84, promoting its localization to the centrosome and facilitating HsSAS-6 accumulation on the centrosome for efficient pre-centrosome assembly and subsequent normal mitosis. However, during late mitosis, NAT10 promotes degradation of HsSAS-6 by acetylating CCDC84 at K31, thus preventing excessive HsSAS-6 from inducing aberrant centromere replication and avoiding abnormal cell division (Wang et al. 2019) (Fig. 5). Furthermore, NAT10 co-localizes with kinesin Eg5 within the centrosome. By acetylating Eg5 at K711, it reduces ubiquitination levels to enhance Eg5 stability and maintain proper loading onto the centrosome (Zheng et al. 2022a, b) (Fig. 5). This process is crucial for Eg5's involvement in bipolar spindle formation by controlling centrosome movement and regulating spindle geometry (Hata et al. 2019).
Fig. 5
NAT10 participates in cellular phenotypes as a protein acetyltransferase. NAT10 catalyzes the protein acetylation-mediated mechanisms of CCDC84, Eg5, H2B, H4, and α-tubulin, promoting cell division. NAT10 catalyzes the acetylation of α-tubulin, leading to a nuclear-cytoplasmic ratio imbalance and causing cell senescence. Similarly, NAT10 catalyzes the acetylation of PARP1, MORC2, and p53, affecting their molecular mechanisms to promote DNA damage repair. Additionally, NAT10 affects cell autophagy by catalyzing the acetylation of Che-1 and NAT10 self-acetylation
In addition to centrosome-associated proteins, NAT10 also targets histone in cell division. Moreover, at the end of mitosis, hsSUN1 induces NAT10 to acetylate histone H2B and H4, restoring highly spiralized chromosomes to chromatin for gene expression preparation during interphase (Chi et al. 2007) (Fig. 5). Therefore, NAT10 plays a significant role in cell division by targeting centrosomes and histones through acetylation modification as well as other pathways (Fig. 5).
Additionally, NAT10 also plays an “important role” in cellular senescence. Larrieu et al. discovered that fibroblasts of HGPS exhibit a more stable microtubule network than normal cells, and the use of remodelin to inhibit NAT10 resulted in decreased microtubule stability, reduced microtubule anchoring and cytoskeletal forces on the nuclear envelope, thereby improving the nuclear shape and adaptability of HGPS cells and reshaping their cellular phenotype (Larrieu et al. 2014). Further studies by the same team revealed that increased microtubule stability in HGPS cells hindered Transportin-1 (TNPO1) entry into the nucleus, resulting in an imbalance of TNPO1 nuclear-cytoplasmic ratio which led to blockage of nuclear transport, gene expression regulation network disorder, and ultimately cell senescence. Remodelin restored the normal TNPO1 nuclear-cytoplasmic ratio, enhanced its nuclear import with carried proteins, restored nuclear pore complex integrity, rebalanced transport, and prevented premature senescence, thereby enhancing the adaptability of HGPS cells (Larrieu et al. 2018) (Fig. 5).
NAT10 participates in the repair of DNA damage as protein acetyltransferaseNAT10 serves as a crucial regulator of p53 homeostasis, employing dual mechanisms of regulation (Liu et al. 2016). In normal circumstances, NAT10 facilitates MDM2 ubiquitination and degradation through its E3 ligase activity at residues 456–558, thereby impeding MDM2-mediated p53 ubiquitination to stabilize p53. Upon DNA damage, NAT10 expression is upregulated and translocates from the nucleosome to the nucleoplasm, directly binding to p53 and preventing MDM2-p53 interaction. Additionally, NAT10 acetylates p53 at K120 to activate it, subsequently promoting cell cycle arrest and apoptosis mediated by p53 in order to maintain genomic stability. This suggests that NAT10 establishes a positive feedback loop with p53 in response to DNA damage (Liu et al. 2016) (Fig. 5). Moreover, NAT10 can bind mutant forms of p53, counteracting MDM2-induced ubiquitination and enhancing the stability of mutant p53 while facilitating proliferation in cells carrying such mutations (Li et al. 2017) (Fig. 5).
The PARP1-MORC2-NAT10 positive feedback loop serves as a crucial regulatory switch for DNA damage regulation. Following DNA damage, activation of PARP1 occurs upon its binding to the fragmented DNA, leading to enhanced interaction with the chromatin remodeling enzyme microrchidia family MORC2. This recruitment of MORC2 by PARP1 facilitates effective DNA repair through catalyzing covalent modification of MORC2 via PARylation within the central region of its CW-ZF domain, thereby promoting chromatin remodeling. Conversely, MORC2 counteracts CHFR protein-mediated ubiquitination and degradation of PARP1 by augmenting NAT10-mediated acetylation at K949 on PARP1, thus stabilizing it (Zhang and Li 2019) (Fig. 5). Further investigations have revealed that DNA damage induces increased interaction between NAT10 and MORC2, resulting in acetylation of MORC2 at K767. K767-acetylated MORC2 binds to histone H3 phosphorylated at T11, facilitating dephosphorylation of histone H3 T11 upon DNA damage and subsequently inhibiting transcriptional activity of CDK1 and cyclin B1 (Shimada et al. 2008; Castedo et al. 2004; Löbrich and Jeggo 2007). Failure in catalyzing acetylation of MORC2 at K767 by NAT10 may impede G2 phase blockade, causing impaired DNA repair prior to entering mitosis and ultimately leading to mitotic abnormalities (Liu et al. 2020) (Fig. 5).
NAT10 participates in cell autophagy as protein acetyltransferaseThe autoacetylation state of NAT10 plays a crucial role in regulating cell autophagy and apoptosis. Under conditions of sufficient energy, NAT10 undergoes autoacetylation at the K462 site, leading to acetylation of the autophagy regulator Che-1 at K228 by NAT10 with autoacetylation at K462. This acetylation event inhibits the transcriptional activation of Che-1-mediated mTOR signaling pathway inhibitors Deptor and Redd1, thereby activating the mTOR pathway to suppress autophagy (Passananti et al. 2007) (Fig. 5). In situations where energy is limited, SIRT1 deacetylates NAT10-K462, resulting in loss of its ability to acetylate Che-1 and inhibit autophagy. Consequently, autophagy is induced as a mechanism for maintaining cell survival during energy deficiency (Liu et al. 2018) (Fig. 5).
NAT10 participates in tumor progression as protein acetyltransferaseNAT10 not only functions as a protein acetylase in normal cells, but also plays a similar role in tumours, promoting tumour proliferation and metastasis.Liu et al. found that in breast cancer (BC) cells, NAT10 acetylates MORC2 at K767 and subsequent binding of acetylated MORC2 to the histone H3 phosphorylation at threonine 11 (H3T11P), which inhibits the transcription of CDK1 and CCNB1, thereby activating the G2 cell cycle checkpoint. This confers resistance to methyl methanesulfonate (MMS) and ionizing radiation (IR) on BC cells, which improves their survival in unfavorable environments and promotes their proliferation and metastasis. In contrast, inhibition of NAT10 by using remodelin or siRNA, sensitivity to MMS and IR can be restored in BC cells (Liu et al. 2020) (Table 1).
Table 1 NAT10 participates in tumor progression as a protein acetyltransferaseIn addition, in Hepatocellular carcinoma (HCC) cell lines, the protein level of NAT10 is positively correlated with that of mutant p53. NAT10 augments the stability of mutant p53 through acetylation and restrains its ubiquitination by impeding the activity of E3 ubiquitin ligase MDM2, thereby promoting the proliferation of HCC cells (Li et al. 2017)(Table 1). Moreover, cytoplasmic localization of NAT10 is associated with a worse prognosis compared to nuclear localization. The worst prognosis is observed in patients with membrane-localized NAT10. This can be attributed to NuLS-induced translocation of NAT10 from the nucleolus to the cytoplasm and cell membrane where it promotes α-tubulin acetylation and stabilizes the tubulin state thereby facilitating metastasis and invasion of liver cancer cells (Li et al. 2017; Zhang et al. 2015) (Table 1).
In addition, Zhang et al. found that cytoplasmic accumulation of NAT10 can enhance α-tubulin acetylation, stimulate F-actin remodeling, and promote metastasis in colorectal cancer (CRC) cells (Zhang et al. 2014).
NAT10 influences cellular phenotypes as RNA acetyltransferaseNAT10 participates in cell division and proliferation as RNA acetyltransferaseThe subcellular localization of NAT10 and its involvement in cell division suggest a pivotal role in cellular proliferation (Shen et al. 2009). Ac4C modification of rRNA is crucial for the biogenesis of 18S rRNA, with NAT10 gene knockout resulting in the accumulation of 30S precursor of 18S rRNA and subsequent growth retardation in human cells (Ito et al. 2014a, b; Drygin et al. 2011) (Fig. 6).
Fig. 6
NAT10 participates in cellular phenotypes as RNA acetyltransferase. NAT10 catalyzes the ac4c modification of mRNAs for CCND2, JUN, and ETV4 genes, which mediates related mechanisms and promotes cell proliferation. NAT10 also catalyzes the ac4c modification of mRNAs for VEGFA, OCT4, SOX2, and RUNX2, etc. genes, which promotes related pathways and affects cell differentiation. Furthermore, NAT10 modifies the ac4c modification of P16 mRNA, which changes the activity of the P16/CDK6/CCND1 signal axis and affects cell senescence
Fei et al. demonstrated that injecting mesenchymal stem cells (mAF-MSCs) derived from amniotic fluid into mice with cryodamage stimulates the corneal microenvironment and enhances corneal endothelial cell proliferation. This effect is mediated by three members of the NAT family, namely NAT10, NAT12, and NAT15. These members enhance the ac4C modification on mRNA associated with CCND2, Jun, and ETV4, which are play a role in regulating cell proliferation within the eyeball. Consequently, this activation triggers the ETV4/JUN/CCND2 signaling axis leading to improved stability and growth promotion of corneal epithelial cells for repairing cold-induced injury in mice(Fei et al. 2021) (Fig. 6). Additionally, NAT10 may contribute to cell proliferation by promoting transforming growth factor beta 1 (TGFB1) expression. Shenshen et al. revealed that chronic exposure to particulate matter smaller than 2.5 µm(PM2.5) increases NAT10 expression levels which subsequently enhance ac4C modification and stability of TGFB1 mRNA. This ultimately results in elevated expression levels of TGFB1 mRNA and protein leading to epithelial-mesenchymal transition (EMT) as well as pulmonary fibrosis development within lung epithelial cells (Shenshen et al. 2023) (Fig. 6).
NAT10 participates in cell differentiation and senescence as RNA acetyltransferaseBy means of mediating ac4C modification of RUNX2 mRNA, NAT10 enhances the up-regulation of RUNX2 expression and transcriptional activity, leading to increased expression of various osteoblast markers such as type I collagen, osteocalcin, or bone sialoprotein. This ultimately promotes osteoblast differentiation in vitro using bone marrow mesenchymal stem cells (BMSCs) (Yang et al. 2021) (Fig. 6). In hPDLSCs, NAT10 acetylates VEGF-A, promoting VEGFA-mediated PI3K and Akt phosphorylation. Subsequently, the PI3K/Akt signaling pathway stimulates proliferation and osteogenic differentiation in hPDLSCs, resulting in ossification (Cui et al. 2023) (Fig. 6). Chen et al. observed that NAT10 is found to be expressed in reproductive organs including the testis, ovary, and epididymis. Conditional inactivation of NAT10 reduces ac4C stoichiometry with a specific decrease in ac4C modification related to spermatogenesis function genes. Transcriptome analysis indicated down-regulation of 381 genes and up-regulation of 662 genes upon deletion of NAT10 which leads to spermatogonia differentiation defects causing male infertility (Chen et al. 2022). Furthermore, mRNAs encoding core pluripotency regulators OCT4 (POU5F1), SOX2, and NANOG along with other pluripotency-related gene transcripts like RTF1, RBL2, JARID2, WDR5, MED1, and EED can be modified by ac4c suggesting a positive correlation between NAT10 expression and cell pluripotency levels. The knockout of NAT10 can significantly diminish the stability of OCT4 mRNA, thereby inducing human embryonic stem cells (hESC) to transition from self-renewal to differentiation (Liu et al. 2023a, b) (Fig. 6). This finding contradicts previous studies where low expression of NAT10 was found to inhibit cell differentiation. It is postulated that this discrepancy may be attributed to variations in cell types, which necessitates further investigation.
In addition, studies have found that NAT10 may also have a biological role in cell senescence. Geng et al. discovered that electroacupuncture (EA) stimulation can suppress the expression of NAT10 in ovarian granulosa cells. This inhibition leads to a reduction in ac4C modification of the cell cycle regulatory protein P16 mRNA, resulting in decreased stability and expression levels. Consequently, it alters the activity of the P16/CDK6/CCND1 signaling axis to inhibit the pathological senescence of ovarian granulosa cells, thereby alleviating follicular atresia and promoting the recovery of ovarian function (Geng et al. 2023) (Fig. 6).
NAT10 participates in tumor progression as RNA acetyltransferaseNAT10 mediates the ac4C modification of the substrate RNA, affecting cell cycle (Geng et al. 2023), mRNA stability (Deng et al. 2023) and translation efficiency (Jin et al. 2022), as well as regulating key processes such as lipid metabolism (Dalhat et al. 2023), thereby promoting the proliferation and metastasis of various tumor cells (Table 2). A large body of studies indicate that intervention in the NAT10-mediated ac4C modification pathway has been shown to have potential therapeutic effects in various tumors, thus providing a new approach for cancer treatment.
Table 2 NAT10 participates in tumor progression as a RNA acetyltransferase NAT10 in esophageal cancerIn addition, NAT10 is upregulated in ESCA and correlates with tumor invasion and metastasis. The utilization of remodelin can effectively inhibit the expression level of NAT10 protein in esophageal cancer cells, thereby impeding the dose-dependent proliferation, migration, and invasion abilities of these cells. This effect may be attributed to the role of NAT10 in mediating ac4C modification on EGFR mRNA. Ac4C modification subsequently enhances translation efficiency, expression levels, and biological activity of EGFR mRNA (Wei et al. 2023). Additionally, NOTCH3, an important substrate for NAT10-mediated metastasis, is found to be significantly upregulated in esophageal cancer tissues. Ac4C modification by NAT10 on NOTCH3 mRNA improves its stability, consequently promoting ESCA metastasis. Knockout experiments demonstrate that elimination of NOTCH3 abolishes the enhancing impact of NAT10 on invasion and metastasis in esophageal squamous cell carcinoma cells (Liao et al. 2023). Long noncoding RNAs(LncRNAs) take part in many physiological and pathological processes through epigenetic, transcriptional, and translational regulations. The ac4C modification mediated by NAT10 enhances the stability of lncRNA CTC490G23.2, leading to a significant upregulation of its expression in ESCA cells and the promotion of tumor metastasis (Yu et al. 2023) (Table 2).
NAT10 in gastric cancerMeanwhile, studies have shown that NAT10 expression is upregulated in gastric cancer (GC) tissues. Zhang et al. discovered that elevated expression of NAT10 enhances the stability of COL5A1 mRNA, an EMT marker, by increasing the ac4C modification level of COL5A1 mRNA. This leads to increased expression of COL5A1 and consequently promotes the occurrence and metastasis of GC (Zhang et al. 2021). Another study revealed that the signal cascade involving the Hp-NAT10-MDM2-p53 axis is implicated in GC development. Specifically, Helicobacter pylori infection induces NAT10 expression, which enhances ac4C modification and mRNA stability of E3 ubiquitin ligase MDM2. Consequently, this upregulates MDM2 expression, which can bind p53, thereby promoting p53 degradation, and contributing to gastric cancer initiation and progression (Deng et al. 2023). Additionally, Yang et al. found that the hypoxic tumor microenvironment facilitates upregulation of NAT10 expression in GC cells through positive regulation by HIF-1α. Upregulated NAT10 then promotes ac4C modification on SEPT9 mRNA. Additionally, a stable protein docking model is formed between SEPT9 and HIF-1α preventing HIF-1α degradation. This establishes a positive feedback loop involving NAT10/SEPT9/HIF-1α which continuously activates downstream glycolysis enzymes within the HIF-1 pathway. As a result, GC cells exhibit enhanced glycolysis addiction in hypoxic conditions leading to improved tolerance towards hypoxia (Yang et al. 2023) (Table 2).
NAT10 in colorectal cancerIn Colorectal cancer, NAT10 binds to the 3'UTR region of KIF23 mRNA resulting in upregulated ac4c modification that enhances KIF23 mRNA stability along with increased translation efficiency for KIF23 protein synthesis. This subsequently activates the Wnt/β-catenin pathway thus promoting CRC development (Jin et al. 2022). Furthermore, Zheng et al. found that NAT10 promotes the stability and expression of Ferroptosis suppressor protein 1 (FSP1) mRNA through ac4C modification. Meanwhile, FSP1 inhibits ferroptosis while promoting proliferation and metastasis in CRC cells. Consequently, this interaction contributes to the progression of CRC mediated by NAT10 activity (Zheng et al. 2022a, b) (Table 2).
NAT10 in cervical cancerIn addition, studies have also demonstrated a significant upregulation of NAT10 expression in cervical cancer (CCa) compared to normal cervical epithelial tissue. This upregulation is positively correlated with poor patient prognosis. Knockout of NAT10 has been shown to inhibit the proliferation, invasion, and migration of CCa cells. Further investigations have revealed that HNRNPUL1 plays a carcinogenic role in CCa. NAT10 can acetylate HNRNPUL1 mRNA through ac4C modification to promote its expression, which in turn accelerates the progression of cervical cancer (Long et al. 2023). Notably, NAT10 may promote cancer cell migration by enhancing tumor tolerance in hypoxic environments through ac4C modification. It was found that NAT10-mediated ac4C modification enhances the translation efficiency of FOXP1 and increases its expression. This activation leads to Glucose Transporter Type 4 (GLUT4) and Ketohexokinase (KHK) transcription in CCa cells, which in turn accelerates glucose uptake and promotes glycolysis and lactic acid accumulation. Ultimately, it promotes Regulatory T cell (Treg) infiltration into the tumor microenvironment resulting in enhanced immune escape for CCa patients thereby promoting the metastasis of CCa cells (Chen et al. 2023) (Table 2).
NAT10 in bladder urothelial carcinomaIn bladder urothelial carcinoma (BLCA), NAT10 plays a crucial role in promoting the proliferation, migration, and invasion of BLCA cells by enhancing ac4C modification on tumor-related mRNAs such as BCL9L, AKT1, and SOX4. This enhancement leads to increased mRNA stability and expression levels (Wang et al. 2022a, b, c). Moreover, Xie et al. found that NAT10 can also safeguard AHNAK mRNA from exonuclease damage by binding to it and stabilizing its structure through ac4C modification. This mechanism enhances DNA damage repair processes which promote cisplatin chemical resistance in BLCA cells. Additionally, it increases sensitivity towards chemotherapeutic drugs after remodelin administration (Xie et al. 2023) (Table 2).
NAT10 in multiple myelomaIn addition, several studies have suggested that NAT10 acts as one of the driving genes in multiple myeloma (MM), promoting both in vivo and in vitro proliferation of MM cells. By utilizing ribo-seq combined with RT-qPCR, researchers identified CEP170, a centrosomal microtubule-anchoring protein, as the primary functional downstream target of NAT10 in MM cells.NAT10 catalyzes ac4C modification on CEP170 mRNA, enhancing its stability and thereby improving translation efficiency. Overexpression of CEP170 stimulates the proliferation of MM cells and induces chromosome instability (CIN) (Wei et al. 2022). Additionally, BCL-XL, an anti-apoptotic protein, serves as another crucial downstream target for NAT10 in MM cells. Through acetylation and stabilization of BCL-XL mRNA, NAT10
留言 (0)