Animals and treatment
Male Balb/c mice (6 ~ 8 weeks) obtained from SPF Biotechnology Co., Ltd. (Beijing, China) were placed in the condition with relative stable temperature (24 ± 2 ℃) and humidity (50 ± 5%) in a 12 h light/dark cycle. The mice were freely obtained food and water. All procedures were conducted according to the guidelines of animal ethics and protocols of Shanxi university of Chinese medicine (AWE202403129).
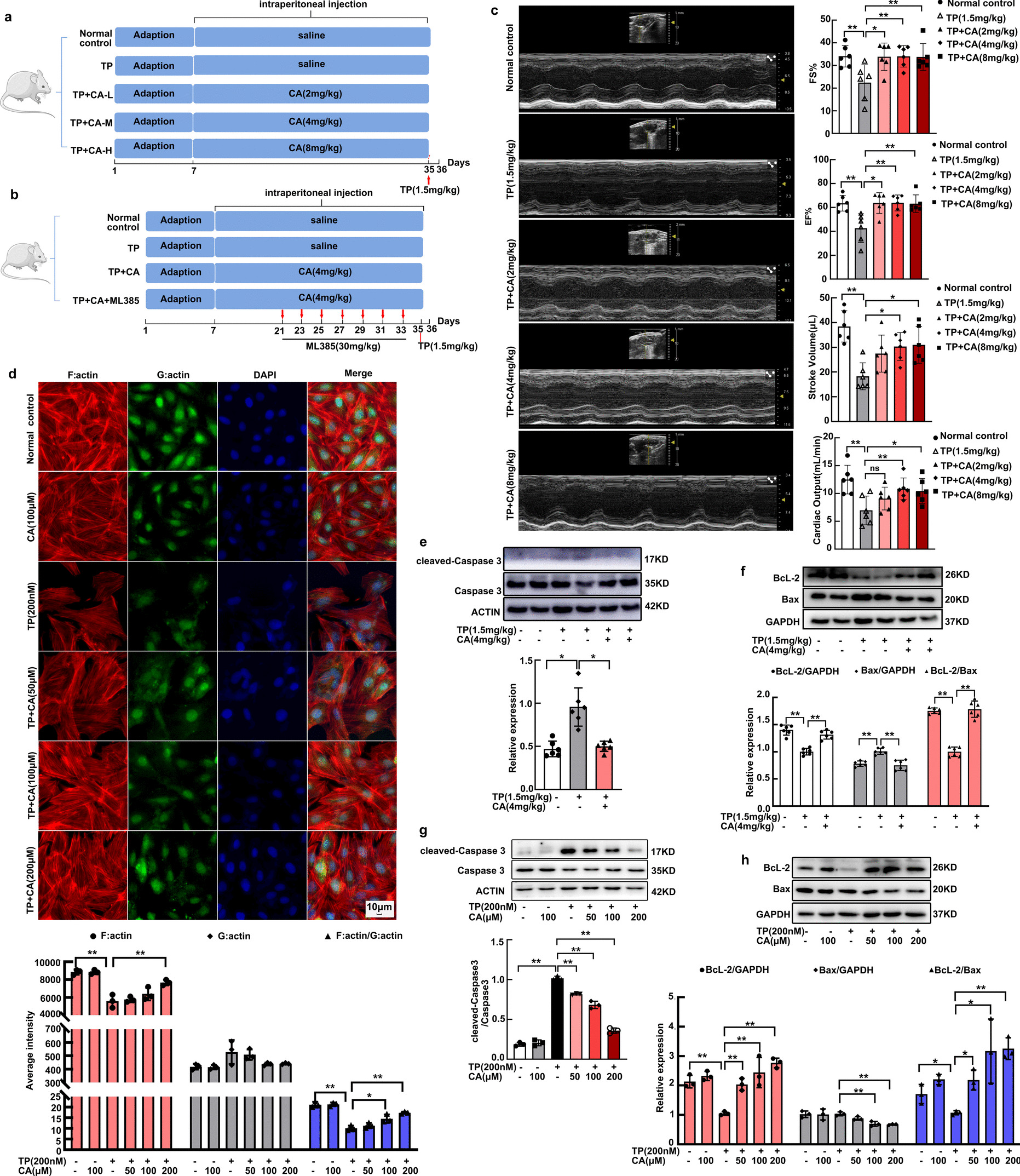
Thirty mice were randomly and averagely divided into five groups with six mice each. The mice were treated with intraperitoneal injection of calycosin once daily for constant 28 days at the concentration of 2, 4 and 8 mg/kg respectively. Following calycosin last injection, triptolide (1.5 mg/kg) was used to establish cardiotoxicity by a single intraperitoneal injection in mice. After 24 h, the mice were anesthetized with isoflurane for cardiac echocardiography determination. Calycosin at 4 mg/kg could significantly protect against triptolide-induced cardiac contraction dysfunction. In the following experiment, 4 mg/kg was chosen for investigating mitochondrial protection of calycosin.
ML385 (# HY-100523, Med Chem Express, USA) was used to investigate the involvement of Nrf2 in calycosin-mediated protection in mitochondria. Twenty-four mice were randomly and averagely divided into four groups with six mice each. Mice were treated with intraperitoneal injection of calycosin (4 mg/kg) once daily for constant 28 days. After the last injection, mice were exposed to a single intraperitoneal injection of triptolide (1.5 mg/kg). ML385(30 mg/kg) were intraperitoneally injected in mice from the fourteenth day of calycosin injection once every two days for consecutive 7 injections. 24 h later following triptolide injection, the mice were deeply anesthetized and their hearts were immediately harvested for mitochondrial respiration detection. The harvested heart was frozen in liquid nitrogen with one part and fixed in 2.5% glutaraldehyde with another part.
Echocardiography
Cardiac contraction was determined by echocardiography in Vero-LAZR-X micro-ultrasound system. Mice were deeply anesthetized in 1.5% isoflurane. Echocardiography of the left ventricle was obtained by slowly adjusting the ultrasound beam. Left ventricular function, including fractional shortening (FS%), ejection fraction (EF%), stroke volume and cardiac output was evaluated from M-mode images.
Transmission electron microscopy
The freshly harvested heart tissues were rapidly fixed in 2.5% glutaraldehyde at 4 ℃ for 2 h and washed with PBS. Hearts were post-fixed in 1% osmic acid at room temperature for 2 h. Following gradient dehydration, tissues were embedded and sliced. The ultramicrocut was stained in 3% uranyl acetate and lead citrate for 15 min and then visualized in transmission electron microscopy (HITACHI, HT7700) to obtain high resolution mitochondrial images.
Mitochondrial respiration detection
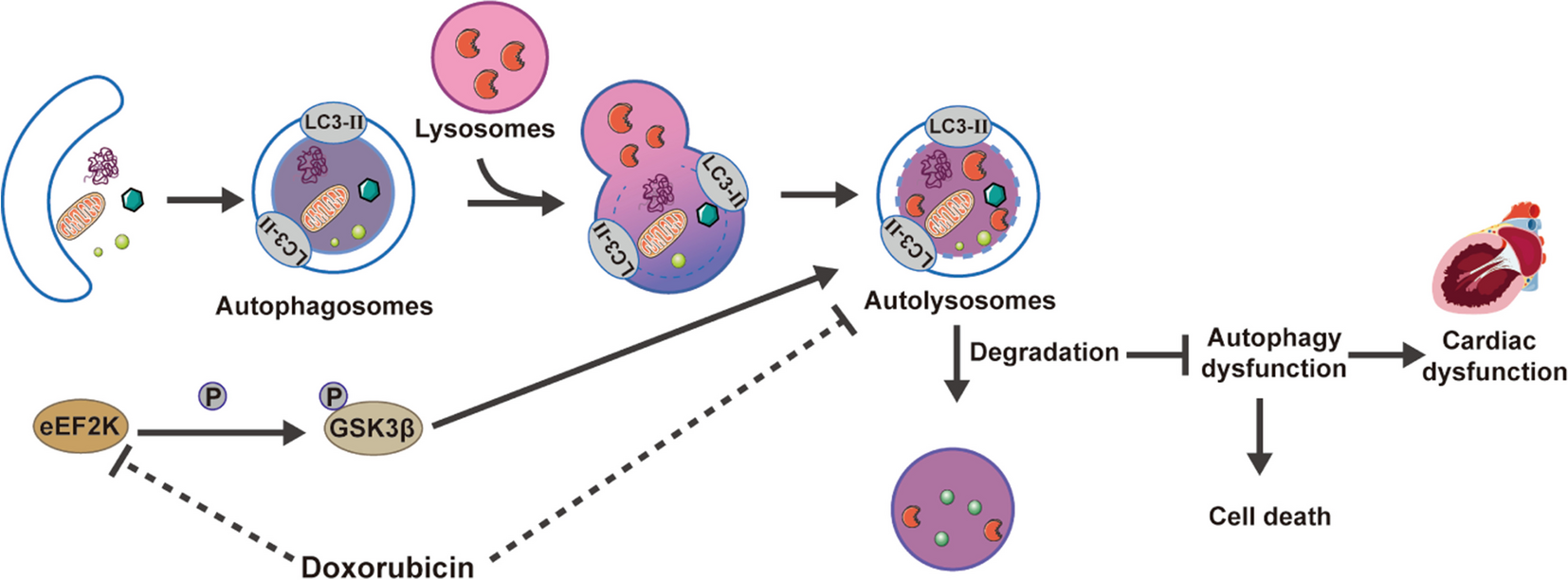
Mitochondrial respiration was detected with high-resolution respirometry (Oxygraph-2 k, Oroboros, Austria). In brief, the freshly harvested heart tissues (10 mg) were homogenized using a Dounce tissue grinder with 200 μL MiR05 (60101–01, Oroboros, Austria). Then 50 µL homogenates were used to determine mitochondrial respiration. Briefly, glutamate (G, 20 mM, Oroboros, Austria), malate (M, 4 mM, Oroboros, Austria) and ADP (D, 1.5 mM, Oroboros, Austria) were sequentially added to determine complex I leak and complex I phosphorylation respiration. Succinate (S, 10 mM, Oroboros, Austria) was used for complex II phosphorylation respiration determination. Oligomycin (Omy, 0.03 µM, Oroboros, Austria), CCCP (U, 0.2–0.3 µM, Oroboros, Austria) and rotenone (Rot, 10 µM, Oroboros, Austria) were respectively used for determining ATP production, maximal uncoupled respiration of electron transfer system and CII ETS. Additionally, antimycin A (AA, 2.5 μM, Oroboros, Austria) was administrated to measure residual oxygen consumption (ROX). Finally, ascorbate and TMPD (As + Tm, 2 mM and 0.5 mM, respectively, Oroboros, Austria) were added to determine complex IV-related phosphorylation respiration. These data were recorded in DatLabacquisition software 5.2 (Oroboros Instruments, Innsbruck, Austria).
Cell culture and treatment
H9C2 cardiomyocytes obtained from “Cell Bank of the Chinese Academy of Sciences” were cultured in DMEM (12100061, Thermo Fisher, USA), consisting of 10% FBS (A5670801, Thermo Fisher, USA), 100 U/L penicillin and 100 mg/mL streptomycin (P1400, Solarbio, China). Triptolide (CAS:38748–32-2, purity ≥ 98%, #A0104, Chengdu Must Biotechnology Co., LTD) (200 nM) treatment for 24 h was used to induce toxicity in H9C2 cardiomyocytes. H9C2 cardiomyocytes were simultaneously exposed to different concentration of calycosin (CAS:20575–57-9, purity ≥ 98%, #A0514, Chengdu Must Biotechnology Co., LTD) (50, 100 and 200 μM) for investigating the protection of calycosin against triptolide-induced cardiomyocyte toxicity.
Cycloheximide (CHX, #HY-12320, Med Chem Express, USA) was used to observe Nrf2 degradation among groups. H9C2 cardiomyocytes treated by CHX (50 μM) were simultaneously interfered with triptolide (200 nM) or triptolide (200 nM) plus calycosin (200 μM) for 0, 2, 4, 8 and 12 h respectively.
MG132 (#HY-13259, Med Chem Express, USA), the proteasome inhibitor, was introduced to investigate the involvement of ubiquitination in the distinct expression of Nrf2 among groups. H9C2 cardiomyocytes with MG132 (10 μM) treatment were concurrently treated with triptolide or triptolide plus calycosin for 24 h.
F:actin and G:actin detection
H9C2 cardiomyocytes were washed and fixed in 4% paraformaldehyde for 10 min, followed by permeabilized with 0.1% Triton X-100 for 5 min. After that, H9C2 cardiomyocytes were simultaneously stained with phalloidin (0.165 μM, 40737ES75, Yeasen Biotechnology, China) and deoxyribonuclease (0.3 μM, D12371, Thermo Fisher, USA) for 90 min at room temperature. DAPI (1:2000, 62249, Thermo Fisher, USA) was used for staining cell nucleus. ImageXpress Micro 4 (Molecular Devices, USA) was used for F:actin and G:actin visualization. The fluorescence intensity of F:actin and G:actin was analyzed and calculated by the software in ImageXpress Micro 4.
Western blotting
Proteins in cardiomyocytes and heart tissues were obtained by RIPA lysis buffer (AR0102, Boster Biological Technology, China) in which 1 mM PMSF (AR1178, Boster Biological Technology) and proteinase inhibitor (04693116001, Roche, Mannheim, Germany) were added. The total protein was harvested by collecting the supernatants after centrifuging lysates at 13,000 × g for 15 min. Protein concentration was determined by BCA kit (AR0197, Boster Biological Technology). Nuclear proteins of H9C2 cardiomyocytes were extracted using the commercial Kit (P0028, Beyotime, China). 20 μg protein were separated using 10% SDS-PAGE, followed by transferring to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad, California, USA). The transferred membrane was blocked in 5% non-fat milk for 4 h at room temperature, and then incubated with specific primary antibody (anti-Bax, 1:1000, 50599–2-Ig, Proteintech; anti-Keap1, 1:1000, 10503–2-AP, Proteintech; anti-ubiquitin, 1:1000, 10201–2-AP, Proteintech; anti-actin, 1:1000, 20536–1-AP, Proteintech; anti-BcL-2, 1:1000, 26593–1-AP, Proteintech; anti-caspase-3, 1:1000, 9662, cell signaling technology; anti-TOM20, 1:1000, 11802–1-AP, Proteintech; anti-NRF1, 1:1000, ab175932, Abcam; anti-Nrf2, 1:1000, 16396–1-AP, Proteintech; anti-TFAM, 1:2000, 22586–1-AP, Proteintech; anti-PGC-1α, 1:1000, PA5-72948, Thermo Fisher; anti-LaminB, 1:5000, A16909, ABclonal; anti-GAPDH, 1:3000, AP0063, Bioworld Technology; anti-HO-1, 1:1000, 10701–1-AP, Proteintech; anti-NQO1, 1:1000, 11451–1-AP, Proteintech) overnight at 4 ℃, followed by incubating with HRP conjugated goat anti-rabbit IgG (1:10,000, 60382121, Thermo Fisher) for 90 min at room temperature. Western blot bands were determined by using ECL kit (RPN2232, Cytiva, USA) on an Amersham Imager 600 (Cytiva).
Immunofluorescence
Immunofluorescence was performed to investigate the expression and visualize the distribution of Nrf2 and TOM20 in H9C2 cardiomyocytes. H9C2 cardiomyocytes were fixed in 4% paraformaldehyde for 10 min and then permeabilized with 0.25% Triton X-100 for 10 min, followed by blocking in 1% bovine serum albumin (BSA) for 1 h. After that, cardiomyocytes were incubated with primary antibodies (anti-TOM20, 1:200, 11802–1-AP, Proteintech; anti-Nrf2, 1:200, 16396–1-AP, Proteintech; anti-Keap1, 1:200, 60027–1-Ig, Proteintech; anti-PGC-1α, 1:200, 66369–1-Ig, Proteintech) overnight at 4℃, followed by incubating with goat anti-rabbit IgG H&L (Alexa Fluor® 488) (1:500, ab150077, Abcam) or goat anti-mouse IgG H&L (Alexa Fluor® 647) (1:500, ab150115, Abcam) for 1 h at room temperature. Cell nucleus was stained by DAPI. The expression of related proteins was visualized using ImageXpress Micro 4 and laser scanning confocal microscope (FV1000, OLYMPUS, Japan).
The expression of Nrf2 and NRF1 in heart tissues were also measured by immunofluorescence. The freshly harvested heart tissues were embedded with OCT, followed by quick-frozen with liquid nitrogen. Heart cryosections at 4 μM thickness were fixed in 4% paraformaldehyde for 10 min, followed by permeabilization with 0.25% Triton X-100 for 10 min at room temperature. Then the cryosections were blocked in 1% BSA for 1 h. After that, the slices were immune-stained with anti-Nrf2(1:200, 16396–1-AP, Proteintech) and anti-NRF1(1:200, ab175932, Abcam) at 4 ℃ overnight. After primary antibody incubation, the slices were incubated with goat anti-rabbit IgG H&L (Alexa Fluor® 488) for 1 h at room temperature. The cryosections were further stained with DAP1 for 5 min to observe cell nucleus. Then the slices were viewed on laser scanning confocal microscope.
ATP measurement in H9C2 cardiomyocytes and heart tissues
H9C2 cardiomyocytes and heart tissues were lysed, and the ATP content was measured using an enhanced ATP assay kit (S0027, Beyotime). The procedure was conducted according to the manufacturer’s instructions.
Quantitative RT-PCR
TRIZOl reagent (108–95-2, TaKaRa, Japan) was used for total RNA extraction. The RNA was reverse transcription to cDNA by TaqMan Reverse Reagents (RR047A, TaKaRa). Quantitative RT-PCR was conducted with a Fast Start Universal SYBR Green Master (RR820A, TaKaRa) on 7900HT Real-Time PCR machine (Applied Biosystems, MA, USA). Gene expressions were measured by 2−ΔΔCt quantification method. Primer sequences were shown in Supplementary Table 1.
Measurement of mtDNA copy number
DNeasy Kit (69504, QIAGEN, Germany) was used to extracted total DNA in accordance with the manufacturer’s instructions. The mRNA expression of cytochrome c oxidase subunit 1 (COX1) that encoded by mtDNA was measured by qRT-PCR, and normalized by GAPDH. The primer sequences were shown in Supplementary Table 1.
Transfection
H9C2 cardiomyocyte with 70–80% confluence was transfected with siRNA for Nrf1, Nfe2l2 and Ppargc1α knockdown respectively by using transfection reagent and related-siRNA (Shanghai GenePharma Co., Ltd). The siRNA sequences were presented below:
siNrf1: 5′-GAGCCACAUUAGAUGAAUATT-3′;
si Nfe2l2: 5′-GGAGGCAAGACAUAGAUCUTT-3′;
siPpargc1α: 5′-GCCAAACCAACAACUUUAUTT-3′;
siNC: 5′-UUCUUCGAACGUGUCACGUTT-3′.
Nrf1 was overexpressed by transfecting pcDNA 3.1-Nrf1 plasmid with Lipofectamine 2000 (11668019, Thermo Fisher, USA) in H9C2 cardiomyocytes. After transfection the cells were cultured in DMEM for 6 h, followed by incubation in complete DMEM consisting of 10% FBS for another 48 h.
Co-immunoprecipitation and ubiquitination assay
Co-immunoprecipitation (CoIP) assay was conducted according to the protocol of a commercial Kit (26146, Thermo Fisher). H9C2 cardiomyocytes were lysed and centrifuged to harvest supernatants. Protein concentration was quantified by BCA kit. Control agarose resin was used to bind 1 mg protein of supernatants to avoid nonspecific binding at 4 ℃ for 2 h. Following that, anti-Nrf2 (1:100, 16396–1-AP, Proteintech) and anti-normal IgG (2729, Cell Signaling Technology) was added for antigen–antibody combination at 4 ℃ overnight. For ubiquitination assay, the incubated antibody was changed to anti-ubiquitin (1:100, 80992–1-RR, Proteintech). The immunoprecipitations were exposed to 20μL protein A + G agarose beads for 4 h. After washing, the beads were then boiled with loading buffer and evaluated by immunoblotting analysis.
Dual luciferase assay
Nrf1 promoter (form −2,000 bps upstream of TSS to 111bps downstream of TSS) was synthesized by Hanbio Biotechnology (Shanghai, China). 293 T cells with the confluence of 70–80% were simultaneously transfected with plasmids of pGL3-basis and pRL-SV40-Renilla by using LipoFiterTM (Hanbio Biotechnology, Shanghai, China), in combination with pcDNA 3.1-Nfe2l2 plasmid or not. After transfecting for 48 h, the lysates were obtained by passive lysis buffer. Dual-luciferase substrate system (Hanbio Biotechnology) was used to simultaneously detect the activities of both firefly luciferase and renilla luciferase.
Statistical analysis
SPSS software (version 22.0) was used to analyze experimental results. Statistical significance between two groups was analyzed by t-test, and one-way ANOVA was used for more group comparison. Data was presented as mean ± standard deviation (SD). P < 0.05 indicates for statistically significance, and P < 0.01 for extremely significance.




留言 (0)