
記住我

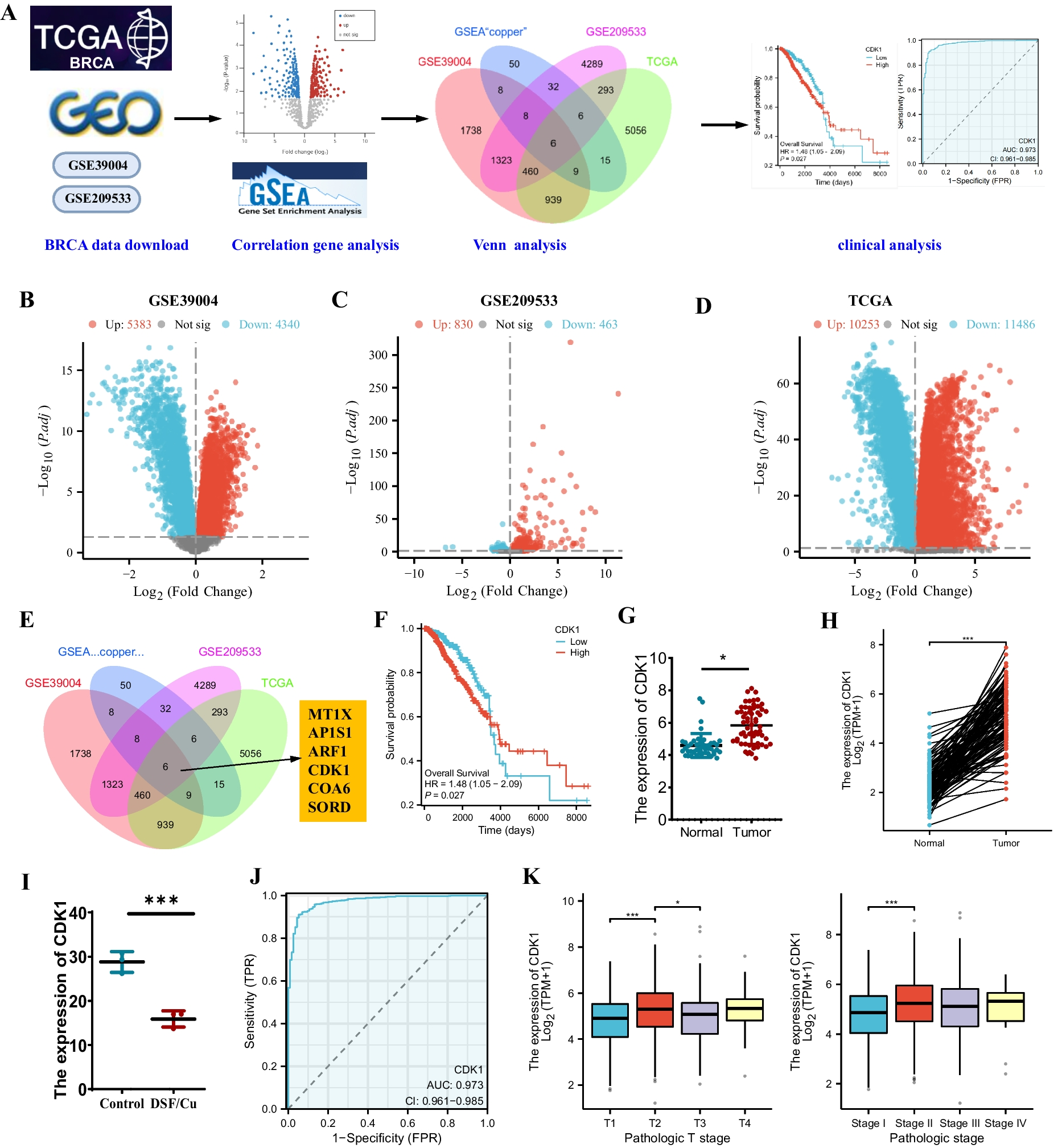


The TCGA database (https://cancergenome.nih.gov/) offers access to the TCGA-BRCA (The Cancer Genome Atlas, TCGA; Breast Invasive Carcinoma, BRCA) dataset for download, comprising mRNA-seq expression patterns alongside pertinent clinical details. The mRNA data encompass 1118 tumor samples and 113 normal samples. Upon downloading the clinical information from the TCGA database, cases with a survival status of 0 or NA (indicating missing survival information and thus excluded from subsequent survival analysis) were omitted, resulting in 970 BRCA patients.
The BRCA RNA-seq dataset GSE39004 includes 47 samples of healthy breast tissue and 61 samples of BRCA tissue. Additionally, the BRCA chip GSE209533, involving copper homeostasis regulator treatment, comprises three groups of DMSO-treated control samples and three groups of DSF/Cu-treated samples. Detailed sample information can be accessed on the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/).
As these data are extracted from publicly accessible databases, ethical clearance or informed consent is not obligatory (Lin et al. 2022).
Acquisition of genes related to copper homeostasisThe Gene Set Enrichment Analysis (GSEA) website yielded 134 genes pertaining to copper metabolism through the extraction of information from 14 pathways associated with copper (Table S1) (Zhang et al. 2021a, b).
Differential expression genes (DEGs) screeningThe GSE45827, GSE139038, and GSE205185 datasets underwent differential analysis employing the "limma" package in the R language adhering to a P-value significance threshold of less than 0.05, resulting in the identification of DEGs. Subsequently, the differential genes were visually represented using the "ggplot2" package to generate a volcano plot (Zhang et al. 2020). The use of the Draw Venn Diagram tool facilitated the performance of a Venn analysis to identify key gene sets from the analysis results (Tan et al. 2023).
Weighted gene co-expression network analysis (WGCNA)Analysis using the "WGCNA" software package was executed on the GSE39004 dataset for WGCNA purposes to explore the biological significance and module information of gene interactions within high-throughput data. The analysis workflow encompassed establishing a gene co-expression network, identifying modules, examining inter-module relationships, and discovering highly correlated genes (Shi et al. 2021). The gene co-expression network establishment process involved the application of a soft threshold to meet the filtering principle of scale-free topology, with soft threshold parameters set at β = 6, scale-free R2 = 0.90, and a cutting height of 0.25 (Pan et al. 2021).
Clinical correlation analysis in the TCGA-BRCA datasetThe TCGA data portal (https://portal.gdc.cancer.gov/) facilitated the retrieval of gene expression data (RNA-seq) and clinical outcome records, in addition to clinicopathological staging information for individuals with BRCA. The levels of CDK1 gene expression and clinical outcome data were extracted. The assessment of ROC (Receiver Operating Characteristic) curves was executed by utilizing the "pROC" package within the R programming platform. Box plots illustrating the differences in CDK1 expression across different T stages (T1-T4) and pathological stages (Stage I-IV) were created applying the "ggplot2" package in R. The Wilcoxon test was applied to analyze the significance of disparities in CDK1 expression levels across various stages.
Reagents and materialsHydrochloric acid (HCl, 10011018), sodium hydroxide (NaOH, 10019762), dimethyl sulfoxide (DMSO, 30072418), toluene (10022818), ethanol (10009218), triethylamine (80134318), benzoyl isothiocyanate (XW05325581), ethylenediamine (10009518), 25% ammonia solution (NH3H2O, 80006765), dichloromethane (DCM, 80047318), sodium sulfate (Na2SO4, 10020518), copper(II) chloride (CuCl2, 10007816), VCR sulfate (VCR, XW20687821), phosphotungstic acid (PTA, XW120679911), and ammonium tetrathiomolybdate (TM, XW1506055601) were all sourced from China National Pharmaceutical Group Chemical Reagent Co., Ltd (Shanghai, China). Trimethoxysilylpropylglycidyl ether (440167) was procured from the Sigma-Aldrich official website. Template silica porous MSN NPs (MSN, 104435, Nanjing Xianfeng Nano Material Technology Co., Ltd). Thermosensitive hydrogel (PLGA-PEG-PLGA) was obtained from Xi'an Ruixi Biotechnology Co., Ltd (R-PLGA-001), and the PHEN GREEN fluorescent probe was purchased from Beijing Bichenglan Biotechnology Co., Ltd (P14313). Throughout the entire experimental process, ultrapure water (18.2 MΩ) produced by an ultrafiltration membrane system (microporous) was used.
Generation and characterization of MBVP NPsThe porous MSN NPs were functionalized gradually on the surface with various chemical substances to generate MBVP NPs.
Synthesis of N-aminoethyl-N'-benzoylthiourea (BTU): After dissolving 1.3 mL of ethylenediamine in 5 mL of dichloromethane and setting it aside, proceed with dissolving 268 μL of benzoyl isothiocyanate in 15 mL of dichloromethane. The two solutions were mixed and added dropwise to the ethylenediamine using a pressure-equalizing funnel. The reaction took place at room temperature (RT) for 4 h, after which a gradual introduction of dilute hydrochloric acid with stirring was conducted until reaching pH = 5 ~ 6. The solution underwent a triple water extraction process, and the final product was obtained after dehydrating the organic phase with anhydrous sodium sulfate and concentrating under vacuum.
MSN-BTU synthesis: 100 mg of MSN was dispersed in 4 mL of toluene, then 2 mL of (3-glycidyloxypropyl)trimethoxysilane was incorporated. The blend underwent a reaction at a temperature of 80 °C for a duration of 48 h, followed by centrifugation, three washes with ethanol, and drying under vacuum for 20 h to obtain epoxy-modified silicon (MSN-EO). Next, MSN-EO was dissolved in 10 mL of dimethyl sulfoxide, 20 mg of BTU, and 20 μL of triethylamine was included, and the reaction unfolded at 40 °C over a period of 24 h. The product was centrifuged, washed with alcohol, and freeze-dried to yield MSN-BTU.
Synthesis of MBVP NPs loaded with VCR: To load VCR into MSN-BTU NPs, VCR (4 mg) was dissolved with MSN-BTU (20 mg) in dimethylformamide (DMF, 1 mL). The blend was agitated under ambient conditions, and phosphate-buffered saline (20 mL) was added. After 6 h of stirring, the resulting mixture was dialyzed in distilled water for 3 days to remove any unbound VCR and DMF solvent, with the external water changed twice daily. Post-dialysis, filtering was carried out on the solution by passing it through a 0.45 μm syringe filter to exclude larger aggregates. The freeze-dried MSN-BTU/VCR material was reconstituted in 4 mL of DMF for UV/Vis spectral evaluation. The VCR loading content was calculated as the ratio of the loaded VCR weight to the weight of the dry sample (Kim et al. 2015).
Following the aforementioned synthesis methods, MBP NPs (MSN-BTU NPs, unloaded VCR), MVP NPs (MSN-EO without attached BTU, loaded VCR), and MBFP NPs (MSN-BTU NPs, loaded with fluorescent marker FITC) were synthesized.
Preparation of MBVP-gel thermoresponsive hydrogelPreparation involved making a 25% aqueous solution of the polymer (PLGA-PEG-PLGA) known for its thermosensitive properties. It remains in a liquid state under low temperatures and transitions to a gel phase at 37 °C. At a low temperature of 4 °C, 5 mg of MBVP NPs was dispersed into 1 mL of the PLGA-PEG-PLGA sol solution. This process was conducted at a low temperature to ensure the sol state remained liquid, facilitating the uniform dispersion of NPs. This step ensured the even mixing of NPs with the thermosensitive hydrogel matrix. After thorough mixing, a thermosensitive nanocomposite hydrogel (MBVP-Gel) at 5 mg/mL concentration was obtained (Rong et al. 2019).
Material analysis and propertiesThe elemental composition across the samples was investigated through scanning electron microscopy (SEM, S-4800, Hitachi, purchased from Shanghai Fulai Optics Technology Co., Ltd.), transmission electron microscopy (TEM) (Hitachi H-7650, purchased from Shanghai Baihe Instrument Technology Co., Ltd.), and energy dispersive spectrometer (Brook QUANTAX EDS spectrometer). Examination of the NPs' morphology was accomplished by TEM (FEI, USA). Fourier-transform infrared spectroscopy (FT-IR, 912A0770, Thermo Fisher, USA) was employed for investigating the chemical composition of the samples. The determination of NPs' size distribution and zeta potential was achieved through the use of a NP tracking analyzer (Zeta View_Particle Metrix, Dachang Huajia Scientific Instrument). Barrett-Joyner-Halenda (BJH) analysis was performed using a physical adsorption analyzer (Norcross, Micromeritics ASAP-2460, USA) to analyze the N2 adsorption isotherm. Utilizing the Brunauer–Emmett–Teller (BET) approach, the specific surface area, total pore volume, and mean pore size distribution curve of the NPs were ascertained.
In vitrodegradation of MBVP-Gel and NPs release experimentThe in vitro degradation of MBVP-Gel was evaluated using digital images and a weighing method. After freeze-drying for 24 h, the initial morphology of the fibers was recorded, and an equivalent weight (W0) was measured. Subsequently, either a PBS solution without or with 20 U/mL lipase was added to small vials containing MBVP-Gel, and these vials were then positioned in a 37 °C shaking incubator at 40 rpm. The solution in the vials was changed every three days. At predetermined time points, the remaining weight of the composite material (W1) was recorded. The equation to determine the remaining percentage of weight can be expressed as: Remaining weight (%) = W1 / W0 × 100% (Chen et al. 2022b, c, a).
To study the release of NPs from the hydrogel, 200 μL of MBFP-Gel hydrogel was placed in 20 mL of PBS and oscillated at 37 °C. At specific time points, 1 mL of the released culture supernatant was taken for fluorescence measurement, succeeding with the inclusion of 1 mL of fresh PBS. Measuring the absorbance at 497 nm (FITC peak) allowed the assessment of the discharged quantity of MBFP NPs.
Simulated physiological environment for VCR release behaviorTo evaluate the release effect of VCR under different physiological conditions using a localized drug delivery system, a pH 5.0 PBS buffer and esterase-simulated lysosomes were used. The extracellular matrix of cancerous tissues was emulated by employing a pH 6.8 PBS buffer, with no esterase component. Additionally, the extracellular matrix characteristic of normal tissues was imitated using a pH 7.4 PBS buffer that did not contain esterase. MBVP NP drug release was assessed at 37 °C in these conditions. The MBVP NP samples were inserted into dialysis pouches (featuring a cellulose membrane of 12 kD molecular weight threshold) and submerged in 50 ml of PBS under diverse pH levels, with or without esterase, and incubated under magnetic stirring (150 rpm) for 120 h. During designated time slots, 2 mL of medium was extracted and substituted with an equivalent amount of fresh PBS. The drug concentration release was measured using a spectrophotometer at predetermined time intervals. Samples were taken at 0, 5, 12, 24, and 48 h (n = 3 for each data point). The accumulated release rate of the drug was evaluated using the mathematical formula: Q = (∑ni = 1 ~ nCi × Vi) ÷ m × 100, where Q denotes the accumulated drug release percentage, Cn is the concentration of the released drug mass, V0 stands for the total PBS volume, Vi represents the volume extracted each time, Ci signifies the drug concentration in the withdrawn volume, and m is the total drug loading capacity (Farasati Far et al. 2023).
Synthesis of a copper ion fluorescent probe (RhB)In accordance with the literature (Gao et al. 2017), Rhodamine B was synthesized. In ethanol solution, dissolve 1.2 g of hydrochloric acid Rhodamine B (2.2 mmol) and stir at RT, then gradually introduce excess hydrazine hydrate (3 ml). Refluxing of the combination occurred at a temperature of 95 degrees Celsius lasting for 4 h, after which it was cooled down in the air and concentrated under pressure reduction, resulting in the formation of an orange solid product. The solid underwent complete dissolution in 1 M hydrochloric acid, and 1 M NaOH was added dropwise until the solution reached a pH of 9–10. The mixture was sieved, rinsed with water, and dried using vacuum, yielding a light pink Rhodamine B solid (Lei et al. 2023).
Response of RhB to copperIn order to enhance the selectivity of Rhodamine B for copper, Rhodamine B (1.5 μM) was mixed with various metal ions (Na, Mg, K, Ca: 5 mM; Zn, Fe, Cu: 10 μM) in a ratio of DMSO: water = 1:10. Subsequently, the fluorescence brightness of Rhodamine B was quantified under excitation at 488 nm. Cu2+ chelates with MBP NPs; hence, Rhodamine B (10 μM) was mixed with 0, 1, 2, 5, and 10 μM Cu2+ in the presence or absence of MBP NPs (30 μg/ml), followed by fluorescence intensity measurement under excitation at 488 nm. Moreover, for fluorescence imaging, the sample treatment steps were identical to those aforementioned and imaging was performed using the IVIS in vivo imaging system (Lei et al. 2023).
Cell culturingTwo cell lines were utilized in this study: MCF-7 (human BRCA cell line, SCSP-531, Chinese Academy of Sciences Cell Bank (CAS Cell Bank)) and 4T1 (mouse BRCA cell line, TCM32, CAS Cell Bank). The cells underwent exposure to escalating levels of VCR (0.05, 0.1, 0.5, 1, 2 μM) for a period of six months to establish drug-resistant variants: MCF-7 drug-resistant cells (MCF-7DR) and 4T1 drug-resistant cells (4T1DR). The cells were cultured in DMEM medium (DF-041, Sigma Aldrich, Shanghai, China) encompassing 10% FBS (A5669701, ThermoFisher), 10 μg/mL streptomycin, and 100 U/mL penicillin (V900929, Sigma, USA) at a concentration of 1 × 105 cells per dish, in a 37 °C humidified incubator with 5% CO2, furnished with 10% FBS and 1% penicillin–streptomycin combination (Farasati Far et al. 2023; Li et al. 2022a, b).
Effect of copper on CDK1 expressionThe MCF-7 cell line was inoculated at a ratio of 1 × 105 cells per well in DMEM medium with the addition of DF-041 (Sigma Aldrich, Shanghai, China) and maintained for 24 h. Subsequently, the cells were treated with 10 μM CuCl2 and 10 μM TM for another 24 h. Cells were subjected to double PBS rinsing post-treatment, and the relative mRNA expression levels of CDK1 were quantified via qPCR analysis, while the relative protein expression levels of CDK1 were examined through Western blot (WB) methodology (Li et al. 2022a, b).
Measurement of copper ion variationTo investigate the impact of drugs on intracellular copper ion levels, cell lysis was initially performed using Triton X-100 (T434386, Aladdin) to release intracellular copper ions. The supernatant was obtained by centrifuging the lysate at 1,000 g for 5 min at a temperature of 4℃. Subsequently, A repeat centrifugation at 1,000 g for 5 min at 4℃ was performed on the supernatant, succeeded by a definitive centrifugation at 12,000 g for 10 min at 4℃. Quantification of copper ion content was carried out by employing Inductively Coupled Plasma Mass Spectrometry (ICP-MS, Agilent 7900, Japan). This study employed ICP-MS technology to measure the levels of copper ions (Yang et al. 2023).
Assessment of cell viabilityCalcein AM (C3099, ThermoFisher, USA) and Propidium Iodide (PI) (P1304MP, ThermoFisher, USA) were utilized for distinguishing between live and dead cells. Initially, cell cultures were treated with Calcein AM (1 μM) and maintained at 37℃ for 30 min, subsequently cleaned with PBS on three occasions. After that, PI (1 μM) was applied for a 10-min incubation, followed by another three rounds of PBS washing. Image acquisition was performed employing a confocal microscope (880, Carl Zeiss AG, Germany), acquiring each picture from a unique viewing area. Three distinct trials were performed for each individual sample. Calculation of cellular viability was executed applying the ImageJ program (V1.8.0) (Liu et al. 2021a, b, c). Additionally, a CCK-8 assay (C0041, Beyotime, Shanghai, China) was performed to assess proliferation rates in chemoresistant and chemosensitive cell groups after 48 h of incubation with VCR treatment. With respect to each trial, 10 μL of CCK-8 detection solution was introduced and underwent incubation for a fixed period of 4 h within the designated cell culture incubation setting. A microplate reader was employed to measure the absorbance at 490 nm, which was utilized for evaluating cell viability (Lee et al. 2022).
MTT assayLogarithmic phase cells were collected, counted, and reconstituted at a density of 5 × 104 cells/mL. Aliquots of 100 μL/well were seeded in 96-well plates, with sterile PBS added to edge wells, and the plate was returned to a 37 °C incubator to allow cells to form a monolayer across the well bottom. After confirming normal cell growth at 24 h, drugs were added according to the experimental protocol. At intervals of 24, 48, as well as 72 h, 10 µL from MTT solution (5 mg/mL; C02, Sigma-Aldrich, USA) was introduced into every well and kept for 4 h. The supernatant was next eliminated, and 100 µL of DMSO was added. Optical density (OD) was checked at 490 nm employing a microplate reader (BioTek, Winooski, VT, USA) to calculate the relative growth rate (RGR) (Li 2024).
IC50 determination of cellsCells underwent cultivation at a density of 1 × 105 cells/well and were exposed to diverse doses of VCR for 48 h (0.05, 0.1, 0.5, 1, 5, 10, 20, 40, 80, 160 μM). Determination of cell viability was conducted via the MTT method. A dose–effect graph was plotted with the logarithm of VCR dosage as the x-axis and cell viability percentage as the y-axis to obtain the IC50 value (Dong et al. 2021; Wang et al. 2017).
Flow cytometry (FCM)Application of FCM was implemented to gauge the extent of cell death. In essence, cancerous cells (1 × 105/well) were initially harvested, cleansed in cooled PBS, and then colored in the dark for 15 min using the assay kit (APOAF-20TST, Sigma-Aldrich, USA). Subsequently, the precipitate was rehydrated in 400 μL binding buffer and dyed with 5 μL Annexin-V given in the package. Lastly, cellular examination was performed with the aid of a flow cytometer. Cells in the upper right quadrant displaying the Annexin V + PI + phenotype indicated late apoptotic cells; those in the lower right quadrant with the Annexin V + PI- phenotype indicated early apoptotic cells; cells in the upper left quadrant with the Annexin V-PI + phenotype indicated necrotic cells, whereas cells in the lower left quadrant with the Annexin V-PI- phenotype represented live cells (Zhou et al. 2021).
Determination of relative gene expression levels by RT-qPCREntire RNA was derived from tissue samples or cellular elements via Trizol reagent (15596026, Invitrogen, USA), and RNA quantity and clarity were measured via NanoDrop LITE (ND-LITE-PR, Thermo Scientific™, Germany) at the wavelengths of 260/280 nm. Subsequently, cDNA was synthesized using the PrimeScript RT reagent Kit with gDNA Eraser (RR047Q, TaKaRa, Japan). The RT-qPCR analysis of genes was carried out applying SYBR Green PCR Master Mix (4364344, Applied Biosystems, USA) and the ABI PRISM 7500 Sequence Detection System (Applied Biosystems).
The primers for each gene were fabricated by TaKaRa (Table S2), utilizing GAPDH as the reference gene. The relative gene expression quantities were computed employing the 2−ΔΔCt methodology, in which the ΔΔCt calculation formula is depicted as: (average Ct value of the target gene in the experimental group—average Ct value of the reference gene in the experimental group)—(average Ct value of the target gene in the control group—average Ct value of the reference gene in the control group) (Ayuk et al. 2016; Wu and Yi 2018; Mao et al. 2019). The RT-qPCR assays were executed in triplicate for each sample.
Cell transfection and groupingTo overexpress the CDK1 gene, we constructed human and mouse CDK1 overexpression plasmids on the gene overexpression plasmid vector pCMV6-AC-GFP (LM-2069, LMAI Bio, Shanghai, China), obtained from Sino Biological (Shanghai, China). Similarly, we constructed human and mouse CDK1-shRNA plasmids applying the pLKO.1-puro vector (QYV0024, Beijing Qiyuan Biotech Co., Ltd., China) with targeting sequences specified in Table S3. Thermo Fisher (USA) provided the non-targeting sh-NC sequences for both human and mouse models. Lentiviruses carrying oe-CDK1 and CDK1-shRNA, abbreviated as oe-CDK1-LTEP-s and sh-CDK1, respectively, were generated on the basis of HEK293T cells (CBP60661, Nanjing KeyBiotech Co., Ltd., Jiangsu, China), leveraging plasmid and lentivirus packaging assistance offered by Sino Biological Engineering. Plasmids harboring a sole luciferase reporter gene were simultaneously transfected with aid plasmids into HEK293T cells employing Lipofectamine 2000 agent (11668030, Thermo Fisher, USA). The lentivirus production involved validation, amplification, and purification of the transfected cells, ultimately leading to packaging.
Culturing 5 × 105 cells in 6-well plates was the initial step for lentivirus-assisted cell transfection, and transfection of the cells took place when they reached 70–90% confluence, utilizing lentivirus (MOI = 10, titer of approximately 5 × 105 TU/mL) and 5 μg/mL polybrene (TR-1003, Merck, USA). Post transfecting for 4 h, an equivalent volume of fresh medium was supplemented to diminish the polybrene concentration. Subsequently, replacement of the media with fresh media occurred at the 24-h post-transfection mark. Transfection efficiency was determined by monitoring the luciferase reporter gene expression after 48 h of transfection, and stably transfected cell lines were obtained by subjecting the cells to puromycin (A1113803, Gibco, Grand Island, NY, USA) selection at a suitable concentration. The retrieval of cells occurred following the ceasing of cell death in the puromycin medium, and validation of knockdown efficacy was established using RT-qPCR methodology.
The groups selected for assessing cell transfection efficiency were as outlined below: (1) MCF-7DR + sh-NC group: MCF-7 drug-resistant cells transfected with non-targeting sh-NC as a negative control; (2) MCF-7DR + shRNA1(Homo) group: MCF-7 drug-resistant cells transfected with shRNA1(Homo); (3) MCF-7DR + shRNA2(Homo) group: MCF-7 drug-resistant cells transfected with shRNA2(Homo); (4) MCF-7DR + shRNA3(Homo) group: MCF-7 drug-resistant cells transfected with shRNA3(Homo); (5) 4T1DR + sh-NC group: 4T1 drug-resistant cells transfected with non-targeting sh-NC as a negative control; (6) 4T1DR + shRNA1(Mouse) group: 4T1 drug-resistant cells transfected with shRNA1(Mouse); (7) 4T1DR + shRNA2(Mouse) group: 4T1 drug-resistant cells transfected with shRNA2(Mouse); (8) 4T1DR + shRNA3(Mouse) group: 4T1 drug-resistant cells transfected with shRNA3(Mouse).
After drug-resistant cell transfection, the following groups were subjected to VCR drug treatment: (1) MCF-7DR + sh-NC + VCR group: MCF-7 drug-resistant cells transfected with negative control sh-NC treated with VCR (IC50); (2) MCF-7DR + sh-CDK1 + VCR group: MCF-7 drug-resistant cells transfected with CDK1 silenced and treated with VCR (IC50); (3) 4T1DR + sh-NC + VCR group: 4T1 drug-resistant cells transfected with negative control sh-NC treated with VCR (IC50); (4) 4T1DR + sh-CDK1 + VCR group: 4T1 drug-resistant cells transfected with CDK1 silenced and treated with VCR (IC50).
Listed below are the experimental groups for the NP treatment rescue procedures: MBVP + oe-NC group: drug-resistant cells transfected with oe-NC plasmid and co-cultured with MBVP; MBVP + oe-CDK1 group: drug-resistant cells transfected with oe-CDK1 plasmid and co-cultured with MBVP.
WB analysisExtraction of total protein from tissues or cells was accomplished with a highly efficient RIPA lysis buffer (C0481, Sigma-Aldrich, USA) encompassing 1% protease inhibitor and 1% phosphatase inhibitor (ST019-5 mg, Beyotime, Shanghai, China), adhering to the guidelines provided by the manufacturer. The lysis process was executed under conditions of 4 °C for 15 min, succeeded by centrifugation at 13000 g for 15 min to collect the supernatant, which was then assessed for protein concentration using a BCA assay kit (23227, TH&Ermo, USA). Subsequently, after quantification with 5 × loading buffer (P0015, Beyotime, China) based on their concentrations, the samples underwent separation via polyacrylamide gel electrophoresis, and were transferred onto PVDF membranes (IPVH00010, Millipore, Billerica, MA, USA). Next, a blocking step was carried out on the membranes using 5% BSA at RT for 1 h before incubating with the primary antibody CDK1 (ab265590, 34 kDa, 1:1000, Abcam, UK) overnight at 4 °C. The membranes were cleansed three times with TBST for 5 min each the following day, then treated with an HRP-conjugated goat anti-rabbit IgG (1:2000, ab205718, Abcam, UK) solution at RT for 1.5 h. Subsequent to the incubation period, the membranes underwent a series of three washes using TBST lasting 5 min each, prior to the application of a chemiluminescent substrate (NCI4106, Pierce, Rockford, IL, USA) to visualize the bands. ImageJ software was utilized for protein quantification analysis, which involved comparing the grayscale values of each protein with the loading control protein β-actin (ab13772, 42 kDa, 1:1000, Abcam, UK) (Wu and Yi 2018). The experiment was replicated thrice to ensure accuracy.
Animal experimentationBALB/c nude mice, specifically five weeks old (strain: BALB/c mice, Beijing Vital River Laboratory Animal Technology Co., Ltd., Catalog No: 211, Beijing, China) were sourced from our institution's Experimental Animal Research Center. The guidelines set by our institution for the ethical treatment and utilization of laboratory animals were strictly followed during all animal studies. The mice were accommodated in a controlled environment with a 12-h light/dark cycle, at a RT of 23 ± 1 °C and a relative humidity of 55 ± 5%, allowing them free access to both food and water. Prior to the commencement of the experiments, the mice underwent a process of acclimatization to these conditions lasting 2 to 3 days (Li and Xiong 2022).
Subcutaneous tumor transplantation experiment in miceValidation Experiment of VCR-Resistant Cell Lines in vivo: Tumor models were established in mice by subcutaneous injection of MCF-7, MCF-7DR, 4T1, and 4T1DR cell suspensions (5 × 106 cells in 100 μL PBS) into the dorsal area. Tumor dimensions were checked every two days for each mouse sample by measuring both length (L) and width (W) using a Vernier caliper, ensuring comprehensive tracking of tumor growth alongside weight assessments. Utilizing the equation V = (W2 × L) / 2 allowed for the calculation of tumor volume (V). Mice were euthanized on the 10th day following tumor implantation, and tumor specimens were extracted to measure copper ion content and CDK1 expression levels (Chen et al. 2023a, b).
Chemotherapy Experiment with Drug-Resistant Tumor Model: The right axillary region of BALB/c nude mice received subcutaneous injections of MCF-7, MCF-7DR, 4T1, and 4T1DR cell suspensions (5 × 106 cells/100 μL PBS) collectively. Seven days later, mice received chemotherapy at three times the IC50 concentration. Tumor samples were collected on day 14 post-treatment, and breast tumor tissues and normal tissues were harvested to measure CDK1 expression levels and copper ion content.
Experimental Treatment of "copper depletion" in Tumor-Bearing Mice: Tumor models were established by subcutaneously injecting 4T1DR cell suspension (5 × 106 cells/100 μL) into the dorsal area of mice, adhering to the ethical standards set by our institution's Animal Experimentation Ethics Committee. The width (W) and length (L) of tumors in every batch of mice were gauged biweekly using a Vernier caliper for monitoring tumor progression and documenting weights. The tumor's volume (V) was determined through the formula V = (W2 × L) / 2.
When tumors reached approximately 100 mm3, mice with tumor burdens were divided into the following groups: 1. Control Group: Intratumoral injection of 100 μL PBS as a positive control. 2. MVP Group: Intratumoral injection of 100 μL MVP for treatment. 3. MBP Group: Intratumoral injection of 100 μL MBP for treatment. 4. MBVP Group: Intratumoral injection of 100 μL MBVP for treatment. 5. MBVP + oe-NC Group: Treatment with intratumoral injection of 100 μL MVP and concomitant tail vein injection of oe-NC lentivirus at a working titer of 5 × 106 TU/mL, with a dosage of 10 μL per mouse, administered continuously for one week. 6. MBVP + oe-CDK1 Group: Simultaneous administration of oe-CDK1 lentivirus through tail vein injection at a concentration of 5 × 106 TU/mL, with a dosage of 10 μL per mouse, given continuously for one week.
Each group comprised 5 mice, and intratumoral drug injections were performed every 2 days. Upon completion of the 28-day duration, the mice underwent euthanasia, followed by dissection and weighing of the tumor tissues (Li et al. 2022a, b).
Tumor tissue stainingTUNEL staining kit was utilized to detect apoptosis in BRCA tissue paraffin sections. Initially, 6 μm BRCA tissue paraffin sections were deparaffinized and hydrated. Subsequently, incubation at RT for 20 min in Tris buffer solution comprising 15.3 mg/mL proteinase K (pH = 8) was conducted, followed by washing with 50 mM TBS (pH 7.6). Next, a reaction with the enzyme solution labeled with green fluorescence (C1086, Beyotime, Shanghai, China) was carried out for TUNEL labeling, culminating in cell nuclei staining with DAPI. Visualization was performed using CLSM (IX73, Olympus Corporation, Japan), followed by image processing and quantitative analysis of TUNEL-positive cells using ImageJ software (Cheyuo et al. 2012; Hu et al. 2021; Su et al. 2018).
Ki67 staining kit was employed to evaluate tumor prognosis in BRCA tissue paraffin sections. Similarly, 6 μm BRCA tissue paraffin sections underwent deparaffinization and hydration. These sections were then incubated at RT for 20 min in Tris buffer solution encompassing 15.3 mg/mL proteinase K (pH = 8), followed by washing with 50 mM TBS (pH 7.6). Subsequent Ki67 labeling was carried out by reacting with the enzyme solution labeled with red fluorescence (AF1738, Beyotime, Shanghai, China), with cell nuclei stained using DAPI. Visualization was conducted using CLSM, followed by image processing and quantitative analysis using ImageJ software (Li et al. 2022a, b).
Hematoxylin and eosin (H&E) stainingTissue damage in breast tumor tissue and normal tissue paraffin sections (heart, liver, spleen, lung, kidney) was examined by means of a H&E staining kit. Initially, obtained tissue samples underwent processing for fixation, followed by deparaffinization of wax-embedded blocks in xylene, hydration in 100% ethanol, 95% ethanol, and 70% ethanol, and finally, mounting or rinsing with water. Hematoxylin dye solution (H8070, Solarbio, Beijing, China) was applied to the designated sections for staining and left for 5–10 min at RT. Subsequently, distilled water was applied to rinse the slides, which were then dehydrated to 95% ethanol and placed in eosin dye solution (G1100, Solarbio, Beijing, China) for 5–10 min. Afterwards, the slides underwent standard dehydration, cleansing, and coverslipping procedures. Eventually, stained samples were viewed through a light microscope to assess staining quality and tissue morphology (Cheyuo et al. 2012; Bian et al. 2021).
Statistical analysisData analysis and bar graph creation were conducted using GraphPad Prism. Descriptive statistics were reported as mean ± standard deviation. Experimental groups were subjected to pairwise comparisons through t-tests. The method employed for multiple group comparisons was one-way analysis of variance (ANOVA) in conjunction with Tukey's post-hoc testing. The appraisal of cellular function at varying time intervals was accomplished utilizing two-way ANOVA, and the assessment of tumor volume was performed by employing repeated measures ANOVA. P < 0.05 was established as the benchmark for statistical significance.
留言 (0)