
記住我
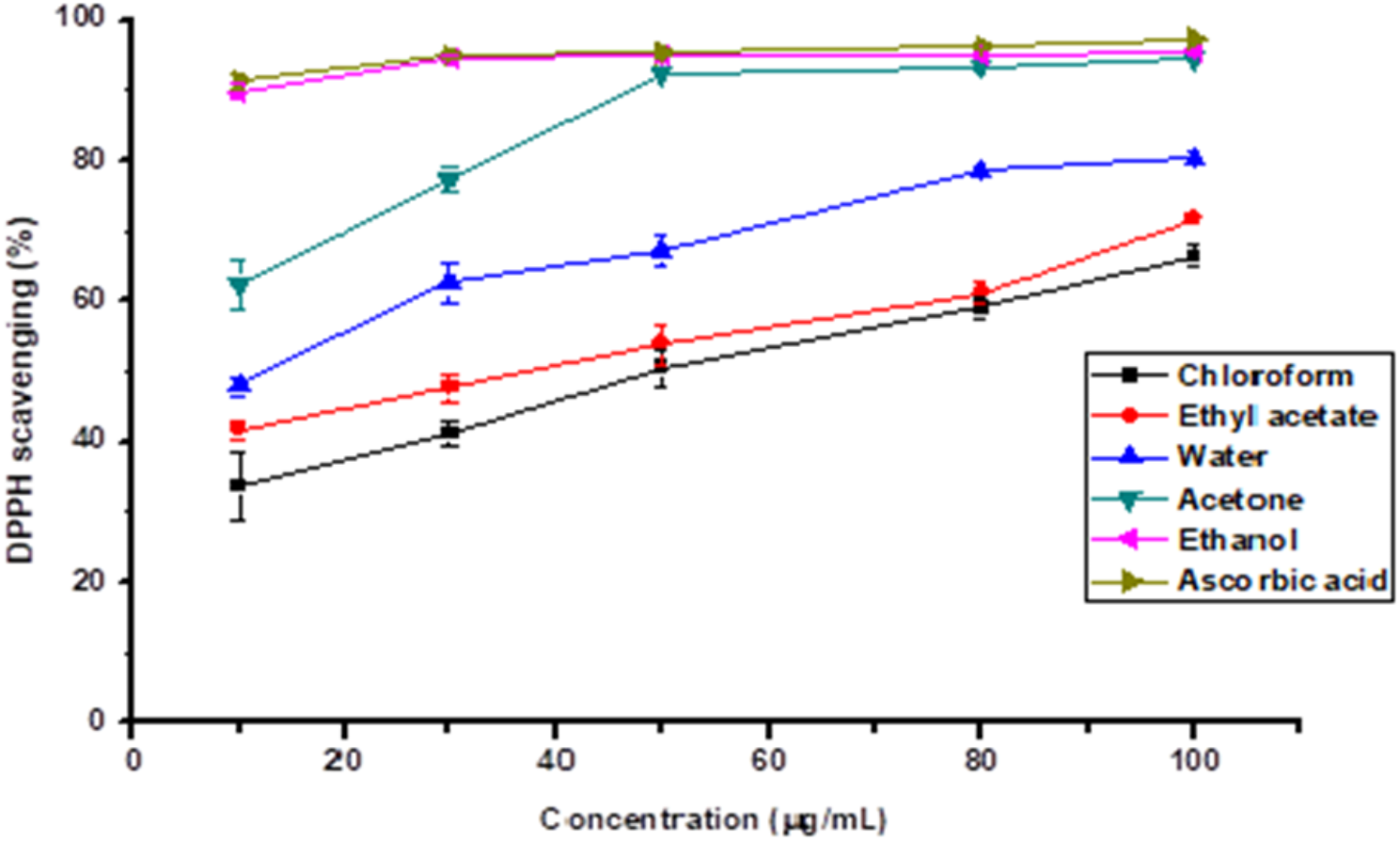
Guang'anmen Hospital (GAMH), China Academy of Chinese Medical Sciences is the initiator and responsible for this programme. Up to now, 13 upper first-class hospitals across 11 provinces in China have participated as subcentres for patient recruitment, diagnosis and treatment, and data collection (Table 1).
Table 1 List of the participating hospitalsEligibility criteria (1)Inclusion criteria
① Patients with advanced CRC confirmed by pathology or cytology and receiving first-line therapy;
② Aged ≥ 18 years old, male or female;
③ ECOG score 0-2 points;
④ Expected survival ≥ 3 months;
⑤ According to RECIST1.1 criteria, at least one detectable lesion;
⑥ Voluntarily join the study, sign informed consent, compliance with good cooperation with follow-up.
(2)Exclusion criteria
① Combined with other malignant primary tumors;
② Immunohistochemistry/polymerase chain reaction/second-generation sequencing results suggest MSI-H/dMMR patients;
③ Patients with recurrence and metastasis within 6 months after radical tumor surgery;
④ Patients who have previously or are undergoing cancer immunotherapy; Patients undergoing radiation therapy;
⑤ Pregnant or lactating women; women of childbearing age and their spouses can not take effective contraceptive measures during and within 6 months after the end of clinical study;
⑥ Psychiatric patients;
⑦ Patients with severe, uncontrolled organic disease or infection, such as decompensated heart, lung, kidney failure caused by intolerance to chemotherapy;
⑧ Patients who have received clinical trials of small molecule drugs within 28 days or received clinical trials of large molecule drugs within 3 months;
⑨ Patients who are known to be allergic to or intolerant of study drugs.
Who will take informed consentPatients with advanced CRC who meet all inclusion criteria and do not meet any exclusion criteria will obtain informed consent. Before each patient is enrolled in the study, a trained investigator will fully and comprehensively introduce the purpose, procedures, and possible risks of the study to the patient or his/her representative and sign a written informed consent form. Patients or their representatives should be informed that their participation is entirely voluntary and they have the right to withdraw from the study at any time. Additionally, it should be clarified to them that their decision to participate or not will have no impact on their regular treatment. During the research process, the personal privacy and data confidentiality of all participants will be protected, and their personal information will not be disclosed.
Interventions Explanation for the choice of comparatorsAccording to NCCN guidelines (Version V1, 2022), the control group receive first-line regimen (FOLFOX/FOLFIRI/CAPEOX ± cetuximab/bevacizumab) in 14–21 day cycles. Specific first-line regimens are shown in Table 2.
Table 2 Chemotherapy regimen ± cetuximab/bevacizumab for advanced CRC Intervention descriptionThe experimental group receive the CKI in addition to the control group regimen. CKI (specifications: 5 ml/tube): ivgtt, 20ml/time, diluted with 200ml sodium chloride injection, qd. The cumulative dose of 200ml should be reached for each cycle of CKI in combination with chemotherapy. CKI is produced and provided by Shanxi Zhendong Pharmaceutical Co., Ltd. (Changzhi, China).
Treatment periodAfter 4–6 months of treatment, patients with complete response (CR), partial response (PR), or stable disease (SD) entered the maintenance phase. The control group receive the maintenance treatment (5-FU/LV/capecitabine ± bevacizumab) in 14–21 day cycles, while the experimental group receive the CKI in addition to the control group regimen. Patients will continue to receive treatment until progressive disease (PD) or an endpoint event. The flow chart of the study design is shown in Fig. 1. If a patient experiences disease recurrence, discontinues treatment due to intolerance, or for other reasons, they will enter a follow-up observation phase, with follow-ups conducted every 3 months until the endpoint event occurs (death or loss of follow-up).
Fig. 1
Flow chart of the study. CKI, Compound Kuhsen injection; PFS, progression-free survival; ORR, objective response rate
Restrictions on concomitant medications and concomitant therapies (1)During the trial treatment, drugs for bone metastases, marrow suppression, nausea and vomiting, diarrhea, abnormal liver and kidney function, infection and other symptomatic treatment may be used in combination, but they should be truthfully recorded.
(2)Immune checkpoint inhibitors are prohibited.
(3)It is prohibited to use traditional Chinese medicines, Chinese patent medicines with anti-tumor effects and other anti-tumor drugs or treatment means other than those specified in the protocol.
Criteria for withdrawals or dropouts from the trial (1)Unexpected events occurred during the treatment and could not adhere to the completion of at least 4 cycles of treatment;
(2)The patient voluntarily withdrew the informed consent form and voluntarily requested withdrawal;
(3)The patient was pregnant or lost to follow-up;
(4)The patient developed poor compliance and was unable to continue the clinical study;
(5)The sponsor terminated the study or administrative authorities withdrew the trial;
(6)Other conditions that the investigators considered necessary to withdraw from the study.
Handling of withdrawals or dropouts from the trialThe investigator is dedicated to ensuring the continuation of proper treatment for each patient unless it is in the best interest of the patient to discontinue participation in the study. Should a patient's participation in the study treatment be terminated, the investigator is obliged to diligently assess the patient's outcomes to the fullest extent possible, ensuring that valuable data is captured and analyzed for the benefit of the research. After discontinuing treatment or withdrawing from the study, patients should enter a follow-up period during which they will undergo periodic follow-ups (once every three months, telephone follow-up is acceptable) to ascertain their survival status, with observations continuing until the occurrence of a terminal event in the patient.
Outcomes (1)Primary outcome
PFS: the time from randomization to tumor progression or death. Patients who have not experienced progression by the cutoff date will have their data censored as of the date of their last follow-up contact.
(2)Secondary outcomes
OS: the time from randomization to death due to any cause.
1-year survival rate: the proportion of patients who survive for more than 1 year from enrollment in the treatment in total patients.
1 year PFS rate: the proportion of the total patients who does not with tumor progression or death within 1 year from enrollment.
ORR: Based on imaging studies, the proportion of patients whose tumor volume reduction has reached the predetermined value and can be maintained for the minimum required duration. Response generally refers to the period from the start of response until tumor progression is confirmed, including CR + PR cases. CR is defined as disappearance of all target lesions and maintained for at least 4 weeks; PR is defined as ≥ 30% reduction in the sum of the long diameters of target lesions and maintained for at least 4 weeks.
Disease Control Rate (DCR): The percentage of patients with advanced cancer who achieve CR, PR, or SD as a result of therapeutic intervention, as determined by imaging studies, including the proportion of CR + PR + SD. SD is defined as the sum of the largest diameters of target lesions does not decrease to the extent of PR, or the sum of the largest diameters of target lesions increases by less than 20%.
Symptoms and quality of life evaluation: Quality of life is evaluated using the Functional Assessment of Cancer Therapy-Colorectal (FACT-C), ECOG scores, and KPS scores. Symptoms of CRC were evaluated using the M.D. Anderson Symptom Inventory.
Safety outcomesAccording to the National Cancer Institute's Common Terminology Criteria for Adverse Events (CTCAE v.5.0), adverse events (AEs) are monitored in patients every two months from baseline to disease progression or death. The incidence rate of drug-related AEs (referring to the proportion of patients who experience adverse events caused by the drugs) will be calculated.
Plans for biological specimensAfter signing the informed consent form, blood samples (8ml), midstream morning urine (15ml) and morning stool (5g) were collected and stored before enrollment, after every 2 cycles of treatment and at the time of disease progression. Blood samples need to be separated into plasma and blood cells. The aliquoted blood, urine, and fecal specimens must be stored in a freezer at −80 degrees Celsius and then centrally transported to Guang’anmen Hospital using a dry ice cold chain. The retention of biological samples will be obtained with the informed consent of each participant. In the current trial, blood samples will be tested for metabolomics and stool samples for gut microbiota and will be used to support research in the future.
Additional consent provisions for collection and use of participant data and biological specimensBeyond the scope of this study, patient medical records, as well as blood, urine, and stool samples, may be utilized in subsequent research endeavors. This potential use is clearly outlined in the informed consent form, which is informed to patients by their physicians. Patient is fully voluntary to sign this statement or refuse.
Participant timelineDuring the screening and enrollment phase, patients are required to complete the collection of demographic information, vital signs, physical examination, performance status assessment, clinical symptoms, and related examinations and imaging tests. Referring to the guidelines, the chemotherapy regimen is set to have a cycle of either 14 or 21 days. All patients in this study will be followed up every 3 months from the end of treatment until the endpoint event (death or loss to follow-up). Patients who discontinue treatment or withdraw from the study should also enter the follow-up period. See Table 3 for the participant timeline.
Table 3 Schedule of screening, intervention and follow-up of the trialSample sizeThe Log-rank test for two groups of survival data is designed with a 1:1 experimental group to control group ratio. According to literature reports, the median PFS for advanced CRC treated with chemotherapy (FOLFOX6 or FOLFIRI) combined with targeted therapy (bevacizumab or cetuximab) is about 8.2 months. Assuming the experimental group has a 4-month longer PFS than the control group, with an alpha level of 0.05, a power of 1-beta (80%), and an expected enrollment time of 24 months for all participants, the expected duration of the clinical trial is 36 months. Using the software PASS 15, it is calculated that 128 cases are needed for both the experimental and control groups. Considering a 20% dropout rate, the final determination is 160 cases for each group, totaling 320 cases.
RecruitmentIn order to complete such large amount patients’ selection as soon as possible, we selected 13 hospitals localized in 11 different provinces in China. In addition, recruitment will be accomplished through posters and website advertisements at outpatient clinics, hospitals. Thus, we will obtain timely enrolment to the greatest extent possible.
Assignment of interventionsAllocation Sequence generationEligible patients will be randomized in a 1:1 ratio to receive first-line chemotherapy ± cetuximab/bevacizumab or CKI combined with first-line chemotherapy ± cetuximab/bevacizumab immediately after signing the informed consent form. The randomization sequence is generated by computer, and randomization will be performed by one physician in GAMH.
Concealment mechanismCentral randomization will be performed using the IWRS system, allocation scheme concealment was implemented, and randomization will be performed using a minimized dynamic randomization method with random factors taking into account: gender (male, female), age (< 60 years, ≥ 60 years), and location (left colorectal, right colon).
ImplementationSteps of central randomization:
(1)Cases will be screened for trial entry based on eligibility criteria by physicians;
(2)The physician will log in the website of central randomization system, enter relevant information (such as patient name abbreviation, age, gender, telephone number, etc.) and click to determine, and the system will generate the grouping results of the case and the corresponding randomization number;
(3)According to the group assignment results, if a participant has been administered the wrong group of medication, no correction will be made and the original drug treatment will continue. Detailed information about the drug treatment will be recorded in the medical records.
BlindingThe design of this trial is open-label, so unblinding will not occur in any circumstances.
Data collection, management, and analysisData collection Plans for data collectionCase report forms (CRFs) will be used to collect data. Within 3 days after the end of the observation course of each patient, the researcher should submit the study medical records, informed consent and other materials to the subject leader of the center for review and archiving. At the same time, the data in the CRFs will be uploaded to the online web-based electronic data capture system for data collection. This data platform features verification and proofreading capabilities, with modified data clearly highlighted in red. Patients’ information on the electronic case report form (e-CRF) will be anonymized using initials and a unique identifier code.
Plans to promote participant retention and complete follow-upData from all participants, including those who discontinue treatment or deviate from the intervention protocol, will be collected faithfully according to the study protocol. Providing blood draw subsidies, transportation allowances, and frequent follow-up phone calls will be conducted to improve adherence to interventions. For patients lost to follow-up, attempts to contact the patient must be made and documented.
Data managementTo ensure that all data recorded in the CRFs are consistent with the source materials, a clinical research coordinator will be assigned to assist the physician in recording and managing all visits and examinations. Each center will be equipped with dedicated file cabinets, with a designated person responsible for the storage of trial documents. At the end of the study, all original data will be kept and reviewed by the study group and archived after the trial is concluded.
ConfidentialityPatient medical records, blood, urine, and stool samples will be anonymized through encoding and encryption before being securely stored at the GAMH. Access to these data and samples will be strictly limited to researchers. We will make every effort to protect the privacy of our patients' personal medical information to the extent permitted by law.
Statistical methods Statistical methods for primary and secondary outcomesSAS 9.4 statistical software will be used by an independent, professional statistician. Full analysis set and per protocol set analysis will be performed simultaneously for efficacy indicators. Safety set analysis will be performed for AEs. All statistical tests will be performed using two-sided tests, and a P value of less than or equal to 0.05 was considered statistically significant. Measurement data shall describe mean, standard deviation, median, minimum and maximum, and enumeration data shall describe frequency and percentage. The use of t-tests and rank sum tests for the analysis of quantitative data, and the use of chi-square tests and Ridit analysis for the analysis of categorical data. The use of Kaplan–Meier method, Wilcoxon rank sum test, or log-rank test for the analysis of survival data.
Methods for additional analysesThe use of the Cox proportional hazards regression model for multivariable survival analysis, with subgroup analysis based on stratification factors (gender, age, and tumor location). At the same time, using PFS as the boundary, while clarifying the survival benefits of CKI in the treatment of advanced CRC, an enrichment design approach is adopted. Through correlation analysis, the advantageous targets of CKI are summarized.
Methods in analysis to handle protocol nonadherence and any statistical methods to handle missing dataAnalyze data for outliers and perform professional analysis of outliers to decide whether to choose them. Data will be analyzed for missing values, and professional analysis will be performed for missing values to decide whether to include dropout or data transfer. The proportion of drop-out cases shall not exceed 20%, otherwise it shall be analyzed and explained.
Plans to give access to the full protocol, participant-level data, and statistical codeThe full protocol, participant-level data, and statistical code during the current study will be available from the corresponding author upon reasonable request.
Oversight and monitoringComposition of the the coordinating centre and trial steering committeeThe trial coordinating centre of the study is established by GAMH and consists of the undertaker and core research members. Investigators from GAMH act as coordinating investigators, tasked with the crucial role of facilitating communication and collaboration between participating hospitals in the trial. They are dedicated to advancing and overseeing the trial's progress, supported by the Contract Research Organization. The trial steering committee consists of the GAMH clinical research team. The committee is responsible for the investigator training, coordination and communication with cooperative units. Periodically hold subject meetings every month to coordinate the progress of the project and solve practical problems in the process of project implementation.
Composition of the data monitoring committee, its role, and reporting structureIn this study, a Data Monitoring Committee (DMC) will be established and an independent expert group with relevant expertise and experience is planned. This committee is responsible for regularly reviewing cumulative data from ongoing clinical statistics in this trial to protect the safety of patients, ensure the reliability of the trial, and the validity of the trial results. If necessary, the DMC may adjust and modify the elements of the ongoing trial based on the collected data, such as intervention dose, study population, or effect size and error for sample size estimation.
Interim analysesThe DMC shall conduct interim analyses based on the progress of the clinical trial to assess efficacy and futility, and continues the trial with modifications to the protocol after the interim analysis, such as adjusting the sample size.
Adverse event reporting and harmsIf any AEs, including serious adverse events (SAEs), occur during the clinical trial, regardless of causal relationship with the CKI, the attending physician should take necessary measures to give treatment and rescue. AEs occurring during the trial should be recorded in the CRF, including the type, severity, time of occurrence, duration, management measures, and the course of management. A comprehensive analysis should be conducted to determine if there is any association with the experimental drug and the control drug being used. After an AE occurs, the investigator may decide whether the patient should discontinue the clinical trial based on the condition. If a SAE occurs during the trial, the investigator should report it to the principal investigator at their respective sub-center within 24 h of becoming aware of the event. The principal investigator of the research unit should then immediately report to GAMH, which will subsequently report to the hospital's ethics committee. Patients discontinued from the trial due to SAE should be followed up until symptoms disappear or treatment is discontinued.
Frequency and plans for auditing trial conductAfter each center completes the enrollment of the first patient, the monitor will conduct one inspection. Following enrollment, each sub-center will be inspected once a month including checking the original data of participants and counting the biological samples. Meet the basic monitoring needs, according to the specific implementation process of the project will be adjusted accordingly.
Plans for communicating important protocol amendments to relevant partiesShould there be any suggested modifications to the protocol, such changes will be thoroughly documented in protocol amendments. These amendments must undergo a rigorous approval process by the ethics committee and regulatory agencies before implementation.
Provisions for post-trial careDoctors and research institutions will do their best to prevent and treat any harm that may result from this research. If a SAE occurs in the clinical study, the medical expert committee will determine whether it is related to the study, and if it is confirmed to be relevant, the research unit will bear the reasonable cost of treatment and the corresponding financial compensation in accordance with Chinese law.
Dissemination plansThis study will be published no matter whether positive or negative results are obtained, and the personal privacy of patients will not be disclosed in the published articles.
留言 (0)