Cell culture and transfection
The human breast cancer cell lines MDA-231 was purchased from the Korea Cell Line Bank (Seoul, South Korea). In contrast, the Hs578T cell line, a TNBC cell line frequently used in studies of aggressive breast cancer phenotypes due to mesenchymal characteristics, was obtained from Woo Keun Song (Gwangju Institute of Science and Technology). MDA-231 cells were cultured in Roswell Park Memorial Institute 1640 medium (RPMI 1640; #31800022; Gibco-BRL, Grand, NY, USA) supplemented with 10% fetal bovine serum (FBS; # US-FBS-500; GW Vitek, Seoul, South Korea), 100 units/mL penicillin, and 100 μg/mL streptomycin (#LS202-02; WelGENE, Daegu, South Korea). Hs578T was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; #12100046; Gibco-BRL) supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin. All cell lines were incubated at 37 °C and 5% CO2 conditions. For transient transfection, 2.5 μg of plasmids were transfected into MDA-231 and Hs578T cells using Lipofectamine 2000 Reagent (#11668019; Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Plasmids construction
End-binding 1 (GFP-EB1) (#KE0822_mNEGFP) and SEC61 translocon subunit beta (pCNS-SEC61B) (#KU001654) were obtained from the Korea Human Gene Bank (Daejeon, Korea). SEC61B was cloned into pEGFP-C1 (#6084-1; Clontech, Palo Alto, CA, USA) and mRFP-C1 (#54764; Addgene, Cambridge, MA, USA). Mito-GCaMP6f and ER-GCaMP6-150 were provided by Sung Hyun Kim (Kyung Hee University). Expression vectors were generated through restriction enzyme digestion. Lentiviral shRNA oligonucleotides were employed, including human ATAT1 shRNA #1 (5′-ACCGCACCAACTGGCAATTGA-3′), and shRNA #5 (5′-AACCGCCATCTTCTTTATATTT-3′) targeting the coding region of ATAT1, human ERN1 shRNA (5′-AGGGCCTGGTCACCACAATTA-3′) targeting the 3′UTR of ERN1, human EIF2AK3 shRNA (5′-TAGCAGCAATCCCTAATATAT-3′) targeting the 3′UTR of EIF2AK3, and human HDAC6 shRNA (5′- CATCCCATCCTGAATATCCTT-3′). These shRNA oligos were cloned into the pLKO.1-blast vector (#26655; Addgene). For ATAT1 overexpression, the coding sequence (CDS) of ATAT1 was obtained by PCR using pEF5B-FRT-GFP-αTAT1 (#20799, Addgene), and cloned into the pcDNA6/myc-His A expression vector (#V22120; Invitrogen). Subsequently, myc-ATAT1 cDNA was generated by PCR from the pcDNA6/myc-His A-ATAT1 construct and cloned into the pLenti-blasticidin vector to create a lentiviral expression vector.
Antibodies and reagents
The following antibodies were purchased from Cell Signaling Technology (MA, USA): Acetyl-α-tubulin (#5335), PERK (#5683), IRE1α (#3294), CHOP (#2895), cleaved PARP (#5625), cleaved caspase-9 (#20750), and cleaved caspase-3 (#9661). Phospho-IRE1 alpha (S724; #NB100-2323) was obtained from Novus Biologicals (Littleton, CO, USA), while phospho-PERK (T982; #ab192591) was purchased from Abcam (Cambridge, USA). Alpha-tubulin (#sc-5286), cytochrome c (#sc-13156), and GAPDH (#sc-32233) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA), and detyrosinated anti-tubulin antibody (#AB3201) was acquired from Millipore (Burlington, MA, USA). α-tubulin (#T9026) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Delta-2 tubulin (#PAB0202) was purchased from Covalab (Villeurbanne, France). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit (#111-035-006), HRP-conjugated goat anti-mouse (#115-035-006), FITC-conjugated goat anti-mouse (#115-095-003), and Cy3-conjugated donkey anti-mouse (#715-165-150) were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Alexa Flour 488 goat anti-rabbit was purchased from Invitrogen (#A-11008). All the antibodies were used for western blotting and immunocytochemical analyses. Eribulin (E7389; Eisai, Tokyo, Japan), tubacin (#537049-40-4; Cayman Chemicals, Ann Arbor, MI, USA and #S2239; Selleckchem, Houston, TX, USA), CCT020312 (#324879; Sigma-Aldrich), thapsigargin (#T9033; Sigma-Aldrich), adenosine 5′-Triphosphate (γ−32P; #BLU502Z500UC; PerkinElmer, Boston, MA, USA), ionomycin, (#I24222; Invitrogen) and MitoTracker Red CMXRos (#9082; Cell Signaling Technology) were used for experiments.
Western blotting
Cells were rinsed with cold phosphate-buffered saline (PBS) and lysed using lysis buffer containing 2% NP-40, 1% sodium dodecyl sulfate, 150 mM sodium chloride, 6 mM sodium hydrogen phosphate, 4 mM sodium dihydrogen phosphate, 2 mM EDTA, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM dithiothreitol, and 1 mM phenylmethane sulfonyl fluoride. The protein lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (#IPVH00010, Millipore). The protein-bound membranes were blocked with 5% skim milk, washed, and incubated with the indicated primary antibodies. Several antibodies were diluted using a SignalBoost Immunoreaction Enhancer Kit (#407207; Millipore) to amplify the antibody reaction. Membranes were subsequently incubated with HRP peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). Protein signals were developed using enhanced chemiluminescence reagents (#1705061; Bio-Rad) and a Fusion Solo S imaging system (VILBER, Collegien, France). Band density was measured using Evolution Capt software.
3-(4,5-Dimethylthiazol-2-yl)−2,5-diphenyl-2H-tetrazolium bromide (MTT) assay
Cell survival rates were quantified using an MTT assay (#MC1029-001–02; Biosesang, Gyeonggi-do, South Korea). Approximately 5 × 103 cells/well were seeded into 96-well culture plates. After incubation, each sample was treated with 500 μg/mL of MTT solution in RPMI or DMEM and further incubated for 2 h at 37 °C. Then, the RPMI medium, including MTT, was removed, and the intracellular purple formazan formed by MTT was solubilized by treatment with dimethyl sulfoxide (DMSO; #DMS555-1; Biopure, Ontario, Canada). The solubilized samples were then analyzed at 570 nm using an Epoch spectrophotometer (BioTek Instruments, Winooski, VT, USA).
Flow cytometry for apoptosis analysis
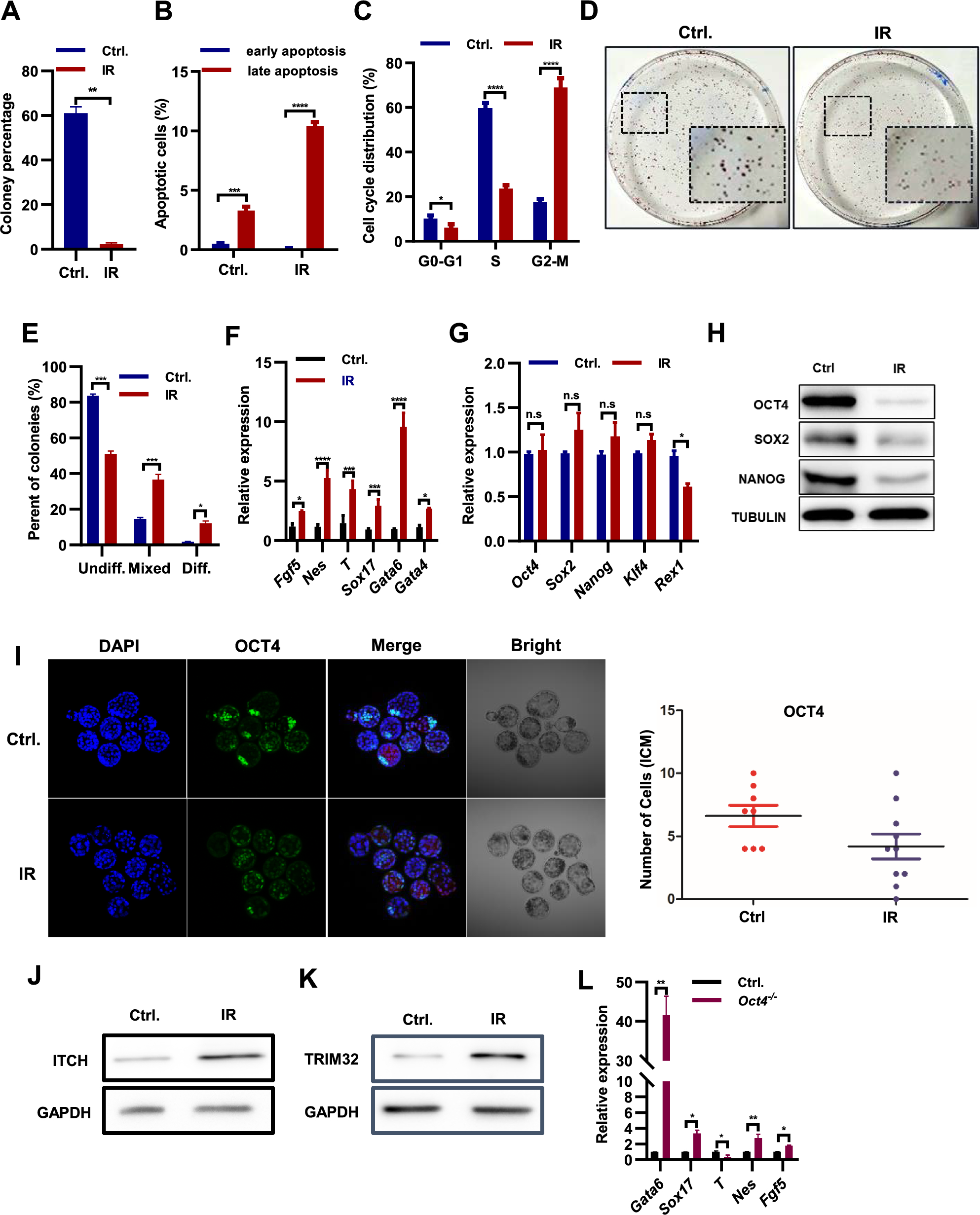
For apoptosis analysis, cells were trypsinized and stained with Annexin V-FITC using the ApoScreen Annexin V Apoptosis Kit-FITC (#10010-02; Southern Biotech, Birmingham, AL, USA) and 7-AAD (#402404; BioLegend, San Diego, CA, USA), according to the manufacturer’s instructions. The stained cells were analyzed using a BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA) and BD Accuri C6 Plus software (BD Biosciences). The percentage of total apoptotic cells was determined as the sum of the percentage of Annexin V+/7-AAD− (early apoptotic) cells and Annexin V+/7-AAD+ (late apoptotic) cells. Additionally, the percentage of living cells was determined using Annexin V−/7-AAD− cells.
Generation of ATAT1 KD and overexpression cell lines
Stable cell lines were generated using the lentiviral vectors. The shRNA- or CDS-cloned constructs were packaged by cotransfection with pMD2.G (#12259; Addgene) and psPAX2 (#12260, Addgene) into HEK293T cells. Lentiviral particles were harvested from HEK293T cells after 72 h and used to infect target cells using 8 μg/mL polybrene. To establish stable cell lines, cells infected with lentiviruses containing shRNA were selected with 1 μg/mL puromycin. In contrast, cells infected with lentivirus containing CDS-cloned constructs were selected with 10 μg/mL blasticidin.
Live cell imaging
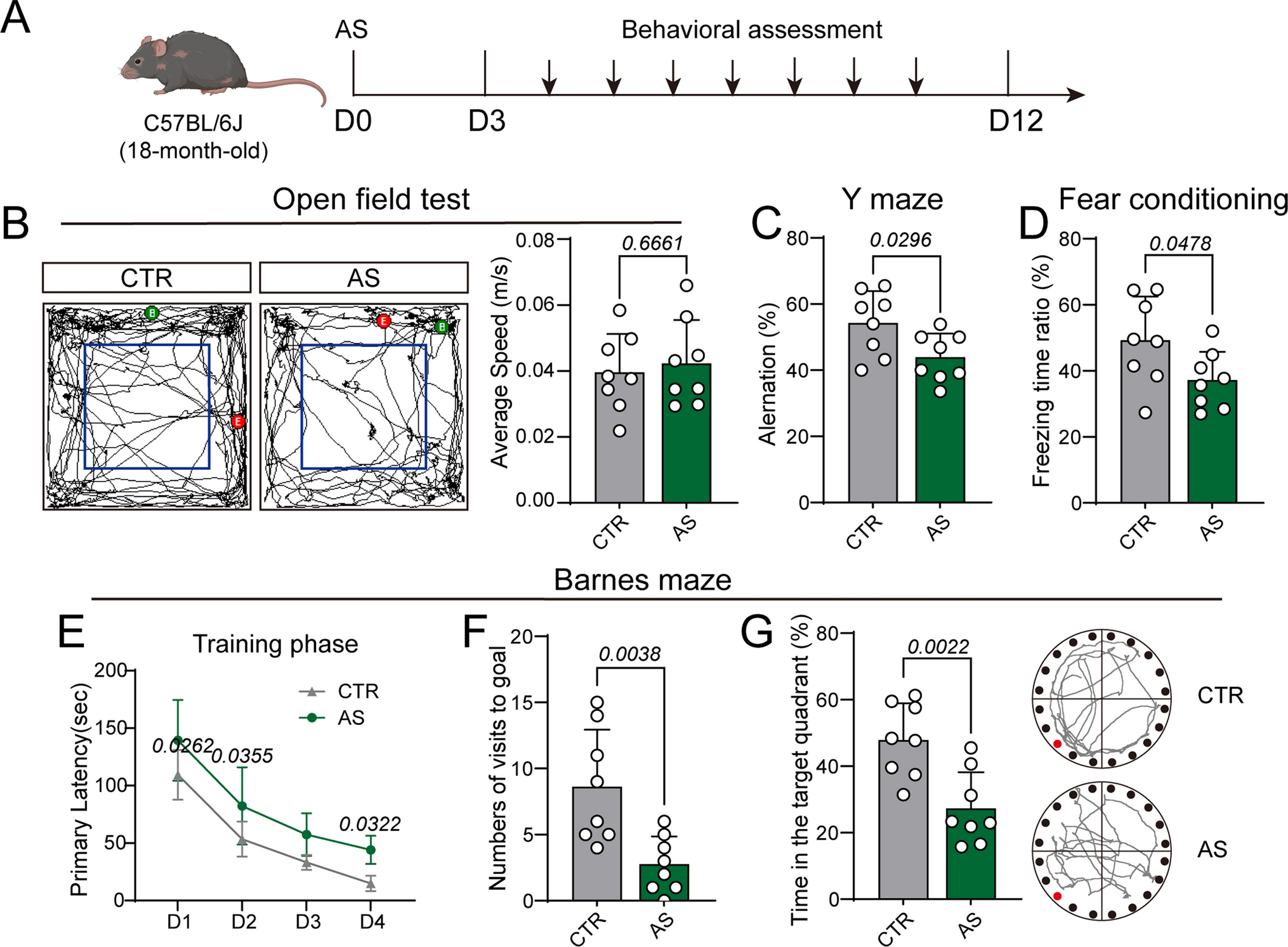
For live imaging experiments, cells were seeded into each well of six-well glass-bottom plates coated with 50 μg/mL rat tail type I collagen and incubated for 24 h post-transfection with plasmids (EB1-GFP for observing microtubule dynamics, and Mito-GCaMP6f and ER-GCaMP6f 150 for observing Ca2+ signal). Next, the cells were treated with or without drugs in FBS-containing growth media in a 5% CO2 chamber at 37 °C. To observe microtubule dynamics, we captured images at 2 s intervals for 30 s using a Nikon ECLIPSE Ti2 inverted microscope system (Nikon, Tokyo, Japan) equipped with a digital camera, DS-Qi2 (Nikon), for live-cell imaging analysis. Microtubule growth rates were assessed by tracking each EB1-GFP comet and measuring its path speed over time using the advanced microscope software NIS-Elements Advanced Research (Nikon).
For observing the Ca2+ signal, cells were washed twice with Hank’s buffered salt solution (HBSS, #NB203-04; WelGENE) and stimulated with ATP (PerkinElmer) and ionomycin (Invitrogen). Images were captured at 200 ms intervals for 5 min using the microscope and digital camera setup described above. The Ca2+ signal in the images was analyzed using the Register ROI plugin (ImageJ) and a time-series analyzer (v3.0).
Immunocytochemistry
Cells were seeded onto 12 mm coverslips coated with 50 μg/mL collagen at a density of approximately 2 × 104 cells/well. After incubation, the cells were fixed with cold methanol for 10 min and permeabilized with 0.5% Triton X-100 in PBS for an additional 10 min. Subsequently, the samples were blocked with 2% BSA and 1% glycine in PBS containing 0.1% Triton X-100 for 1 h. The resulting samples were stained with primary antibodies for 1 h at 22 °C and then incubated with fluorescein-conjugated secondary antibodies for 1 h at 22 °C after washing with 0.1% Triton X-100 in PBS. After staining, samples were mounted using Fluoromount-G (Southern Biotech). Cells were observed using an Eclipse 80i fluorescence microscope (Nikon), and images were captured using a digital camera, DS-Qi2 (Nikon). To quantitatively analyze the perinuclear Mito index and Mito-GCaMP6f signal, we adopted the methods described by [26] and [27], respectively. Images were processed using advanced microscope software NIS-Elements Advanced Research (Nikon) and Photoshop (Adobe Systems, San Jose, CA, USA).
Field-emission transmission electron microscopy
Cells were fixed with 2% glutaraldehyde (#G6257; Sigma-Aldrich) and 2% paraformaldehyde (#G6148; Sigma-Aldrich) in 0.05 M sodium cacodylate (#C0250; Sigma-Aldrich) buffer for 2 h at 22 °C and incubated for 16 h at 4 °C. The samples were washed with 0.05 M sodium cacodylate buffer and incubated in 1% osmium tetroxide diluted in 0.1 M sodium cacodylate buffer for 1 h at 4 °C. The samples were stained with 0.5% uranyl acetate for 16 h at 4 °C after washing with distilled water three times. Next, the samples were dehydrated with 30%, 50%, 70%, 80%, 90%, and 100% ethanol and embedded in Spur’s resin. The samples were sectioned using an ultramicrotome. The samples were stained with uranyl acetate. The images were obtained using a JEM-F200 transmission electron microscope (JEOL, Tokyo, Japan).
RNA sequencing and bioinformatics analysis
Total RNA was extracted from MDA-231 WT, ATAT1 knockout (KO), and eribulin-resistant cell lines. RNA quality and quantity were measured using an Agilent 2100 Bioanalyzer (Agilent Technologies, Amstelveen, The Netherlands) and Nanodrop ND-2000 (Thermo Scientific), respectively. To test and control RNA, we constructed a library using Quant-seq 3′ mRNA-Seq Library Prep Kit (Lexogen, Vienna, Austria), according to the manufacturer’s instructions as previously described [6, 28]. Single-end 75 RNA sequencing was performed using NextSeq 500 (Illumina, San Diego, CA, USA) at Ebiogen Inc. (Seoul, Korea). Functional annotation analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov). Gene ontology (GO) analysis was performed using the ExDEGA GraphicPlus and Revigo software (http://revigo.irb.hr/). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed using ShinyGO 0.77 (http://bioinformatics.sdstate.edu/go/) software. The expression profile was obtained from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). Accession number GSE50811 was used for the GEO analysis.
Three-dimensional (3D) invasion assay
To generate tumor spheroids, 5 × 103 cells were seeded in 96-well ultralow attachment plates (SPL 3D™ Cell Floater, SPL Life Sciences, Gyeonggi-do, Korea, #39724) and incubated with culture media supplemented with 10% FBS for 24 h. Following incubation, the tumor spheroids were coated with a 200 μg/mL growth factor-reduced Matrigel solution for 24 h. As previously described, the tumor spheroids were mixed with 1 mg/mL rat tail type I collagen solution and polymerized for 1 h in a 37 °C incubator [29]. The tumor spheroids embedded within the 3D collagen matrix were then incubated under the specified conditions for 72 h. To analyze the survival of invasive cells under each condition, we stained the tumor spheroids using the Live/Dead™ Viability/Cytotoxicity Kit (Invitrogen, #L3224) following the manufacturer’s instructions. Images were captured using a digital camera (DS-Qi2, Nikon), and cell invasiveness was determined by subtracting the area of invading spheroids from the initial spheroid area using NIS-Elements image analysis software (Nikon).
Live/dead assay
The Live/Dead™ Viability/Cytotoxicity Kit for mammalian cells (#L3224; Invitrogen) was used to evaluate cell viability. Cells seeded onto 12 mm coverslips coated with 50 μg/mL collagen were incubated with 1 μM Calcein-AM and 4 μM EthD-1 for 30 min at 37 °C. After incubation, PBS solution was added to clean the glass slides, and the coverslips were mounted on the glass slides. Images were obtained using an Eclipse 80i fluorescence microscope (Nikon) and captured using a digital camera, DS-Qi2 (Nikon). Images were processed using advanced microscope software NIS-Elements Advanced Research (Nikon, Tokyo, Japan).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
TUNEL assay was performed to detect apoptotic cells using DeadEnd™ Fluorometric TUNEL System (Promega, Madison, WI, USA) following the manufacturer’s instructions.
In vivo xenograft
MDA-231 parental and EriR cells (1.5 × 107) were resuspended in 100 μL of RPMI 1640 without serum admixed with 100 μL Matrigel (#356231; Corning, NY, USA) and injected subcutaneously into six-week-old BALB/c-nude mice (Nara Biotech Co.). Tumor size was measured using calipers every 2 d, and the volume was calculated using the formula L × W2 × 0.5, where L and W represent the tumor length and width, respectively. When the average tumor volume reached approximately 150 mm3, the mice were treated with 0.1 mg/kg eribulin every 2 d through intravenous injection (i.v.) for 14 d and 0.5 mg/kg tubacin every day through intraperitoneal injection (i.p.) or 2 mg/kg CCT020312 every four days through i.p. for 26 d. The tumors were dissected and weighed at the endpoint. The experimental procedures were approved by the Institutional Animal Care and Use Committee of Chung-Ang University (Approval ID: 2019-00133).
Immunohistochemistry (IHC)
Tumors were fixed in 10% neutral buffered formalin (#010–1406-1010; GD CHEM, Chuncheongbuk-do, South Korea) and embedded in paraffin blocks. Sectioned tumors were blocked with an M.O.M™ blocking solution. Sectioned tumors were stained with specific antibodies for acetyl-α-tubulin and cytochrome c. Nuclei were counterstained with DAPI. Sectioned tumors were mounted using Fluoromount-G. Fluorescence-positive cells (%) were analyzed using NIS-Elements advanced imaging software (Nikon).
RNA isolation and quantitative reverse transcription PCR (qRT-PCR)
Total RNA was isolated using RNAiso Plus reagent (#9109; TaKaRa, Tokyo, Japan) following manufacturer’s instructions. For synthesis of complementary DNA, a total 1 μg of RNA was taken with 100 mM oligo primers and PrimeScript reverse transcriptase (#2680; TaKaRa). qRT-PCR was performed with SYBR Premix Ex-Taq II (#RR820; TaKaRa) using QuantStudio 3 (Applied Biosystems, city, CA, USA). The primers used for qRT-PCR are as follows: ATAT1 (forward: 5′-TTTGCATCCTGGACTTTT-3′, reverse: 5′-TTGTTCACCTGTGGGACT-3′), HDAC6 (forward: 5′-AAGTAGGCAGAACCCCCAGT-3′, reverse: 5′-GTGCTTCAGCCTCAAGGTTC-3′), and GAPDH (forward: 5′-GACCCCTTCATTGACCTC-3′, reverse: 5′-TCCTGGAAGATGGTGATG-3′).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA). The significance of differences between data was examined using Student’s unpaired t-test to compare two groups and one-way analysis of variance (one-way ANOVA) or two-way ANOVA to compare more than two groups. Data are presented as mean ± standard deviation (S.D.) from two or three independent experiments. Tukey’s multiple comparison test was performed as a post-hoc test for all one-way ANOVA or two-way ANOVA. One-way ANOVA F values are presented in the figure legends as F (DFn, DFd), where DFn is the df numerator and DFd is the df denominator. p-values less than 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001).

留言 (0)