Patients and samples
In total, 115 peripheral blood samples were collected, including 65 from control patients and 50 from HSV-1-positive patients with lung injury. Sixty-five healthy individuals and 50 patients with HSV-1 infection who met the inclusion criteria were selected. All participants provided their informed consent. Blood samples from the periphery were obtained in the morning while fasting and treated with EDTA anticoagulant to ensure that the blood did not clot. Peripheral blood mononuclear cells were isolated via density gradient centrifugation, washed, and impurities were removed. PBMCs were counted via the cell counting method, and cell activity was detected to ensure high-quality cells. FcRn expression was detected using RT-qPCR. The study received clearance from the Ethics Committee and Institutional Review Board at the First Affiliated Hospital of Xinxiang Medical University, with the approval number EC-024-345.
Cell culture
A549 (human non-small cell lung cancer), 293 T (human embryonic kidney), and African green monkey kidney (Vero) cells were purchased from the Stem Cell Bank of the Chinese Academy of Sciences. A549 cells were cultured in F12 medium (G4560-500; Servicebio, Wuhan, China) supplemented with 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 U/mL) (C100C5; NCM Biotech, Suzhou, China). BEAS-2b, 293 T, and Vero cells were cultured in DMEM (G4511-500; Servicebio, Wuhan, China) supplemented with 10% fetal bovine serum (04-121-1A; Biological Industries, Israel), penicillin (100 U/mL), and streptomycin (100 U/mL) in Dulbecco's modified Eagle's medium (DMEM). The cells were incubated in a 37 °C incubator with 5% CO2 under humid conditions. HSV-1 KOS (GenBank: JQ673480.1) was propagated in Vero cells and titrated as described previously [26]. Viral supernatants were collected and stored at −80 °C.
Mouse models of lung injury
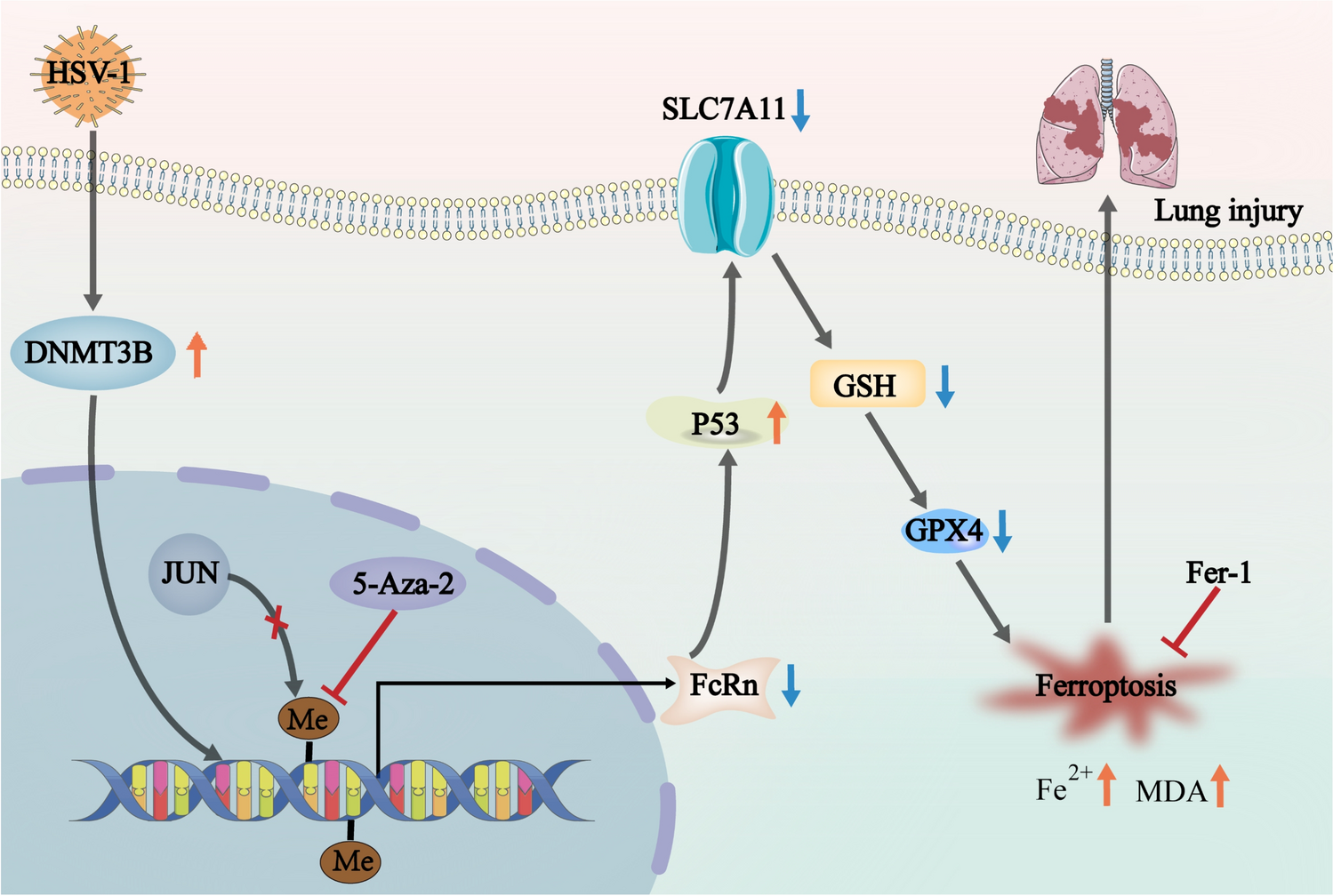
Female wild-type C57BL/6 mice (6–8 weeks old) were purchased from Skibbes (Henan, China). FcRn−/− mice were purchased from Cyagen (USA). FcRn−/− mice used in a previous study were obtained by pairing FcRn heterozygous mice (FcRn±) and genotyping them. Specifically, we confirmed the FcRn−/− genotype via PCR and genomic analysis of heterozygous mice and used them for subsequent experiments (Fig. Sl). The mice were housed in our laboratory animal facility at a room temperature of 20–24 °C and humidity of 50–60%, following a standard 12-h light–dark cycle. All the animals had ad libitum access to food and water. Mice were divided into the following treatment groups: mock, 5-Aza-2, HSV-1, 5-Aza-2 with HSV-1, Fer-1, Fer-1 with HSV-1, FcRn−/−, and FcRn−/− with HSV-1 (n = 6). 5-Aza-2 was purchased from MCE (HY-A0004), and Fer-1 was purchased from MCE (HY-100579). The mice in the infected group were anesthetized via an intraperitoneal injection of zotile (1:7 saline), and the mice were intranasally administered 100 μL of HSV-1 (1 × 105 TCID50/ml). The mice in the 5-Aza-2 treatment group (5 mg/kg/d) and Fer-1 treatment group (0.8 mg/kg/d) received intraperitoneal treatment for 4 consecutive days, starting on the third day after HSV-1 infection. Daily changes in body weight were recorded, and on day 7, the mice were euthanized to remove lung tissues for further experiments. Sterile saline was administered to the rats in the control group. The animal experiments conducted were in compliance with the standards for the treatment and care of laboratory animals, and had received approval from the Xinxiang Medical University Ethics Committee, China (Approval Number XYLL-20230134).
Genomic DNA isolation and bisulfite sequencing
A549 cells were treated with a control medium (no treatment) or medium containing substance1 MOI of HSV-1 for 48 h. Genomic DNA was extracted and subjected to bisulfite treatment using an EpiTect bisulfite kit to convert unmethylated cytosine to uracil, following the manufacturer's instructions. According to the methods in the literature, PCR products from bisulfite-treated genomic DNA were analyzed by pyrosequencing to quantify the site-specific methylation levels. Sequencing samples were processed using a Vacuum Prep workstation (Biotage AB, Uppsala, Sweden). Pyrosequencing was performed using the PyroMark Gold Q96 Reagent and PyroMark ID System (Qiagen, Hilden, Germany) [27]. The primer sequences are listed in Table 1. PyroQ-CpG™ software v. 1.0.9-guided assay design for optimal nucleotide addition order and automated methylation analysis. Pyrosequencing was performed by Sangon Biotech (Shanghai, China).
Table 1 Primers used in bisulfate-sequencing PCRTranscriptomic analysis
Total RNA was isolated from mouse lung tissue via the TRIzol reagent, as described in previous studies [28]. Volcano plots depict the differential gene expression, with each dot representing a gene. The colors and positions of the dots indicate different attributes. The statistical criterion of P < 0.05 was set for significant differences. Partial differential gene expression is shown in the heatmap, with differences indicated based on strict criteria (log2FC > 2, P < 0.05) via DESeq2 identification. The overlap between HSV-1 infection-induced lung injury and ferroptosis has enabled the identification of potential therapeutic targets. The STRING database was utilized to construct protein–protein interaction (PPI) networks for the genes of interest. A confidence threshold > 0.4 was used for the networks. In the PPI networks, nodes represent proteins, node sizes indicate the intensity of the effect on the disease, and internode lines denote the interactions between proteins. Gene set enrichment analysis (GSEA) maps were plotted via the MSigDB database to assess the significance of the ferroptosis-associated gene set, which was performed by calculating the enrichment score (ES) and p-value of the gene set. The foundational data for this research are available in the public repository at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1117674/.
Histopathological lesions
Lung tissues were soaked in 4% paraformaldehyde solution and fixed for 48 h. Then, the tissues were deparaffinized, dehydrated, paraffin-embedded, and cut into 5 µm thick sections. Hematoxylin and eosin (H&E) staining (G1120, Solarbio, China) was performed to evaluate histopathological changes, and a scoring system ranging from 0 to 4 was used. The scores were as follows: 0 = no injury; 1 = mild lung injury characterized by mild bleeding; 2 = slight lung injury with hemorrhage, congestion, and a small amount of epithelial cell detachment as the main features; 3 = moderate lung injury with hemorrhage, congestion, and a large amount of epithelial cell detachment as the main features and mild inflammatory cell infiltration as the predominant features; and 4 = toxic lung injury with hemorrhage, congestion, massive epithelial cell shedding, and massive inflammatory cell infiltration as the predominant features. The extent of lung injury was assessed using ImageJ software.
Immunohistochemistry (IHC)
The paraffin sections were rehydrated by dewaxing, and antigen retrieval was performed at room temperature. The sections were washed with 1 × phosphate-buffered saline (PBS) on a shaker. The cells were incubated with 3% hydrogen peroxide for 10 min at room temperature and washed with PBS. Primary antibodies against GPX4 (1:200, GB124327, Servicebio, China) and FcRn (1:200, DF12607, Affbiotech, USA) were added dropwise and incubated overnight at 4 °C. After washing with PBS, the secondary antibody was blocked, and the cells were incubated at 37 °C for 20 min. After another round of washing with PBS, DAB (DA1010; Solarbio, China) color development solution was added. Finally, the sections were re-stained with hematoxylin, dehydrated, and observed under a microscope.
Western blotting
Lung tissues were ground via a grinder and the treated cells were collected, washed with PBS, and lysed with RIPA buffer (NCM Biotech, WB6501, China) containing a mixture of protease inhibitors (MCE HY-K0010). The protein concentration was determined using a BCA Protein Measurement Kit (Beyotime, P0012S, Shanghai, China). Equal amounts of protein were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5% skim milk powder for 2 h at room temperature, washed, and incubated overnight at 4 °C with the indicated antibodies. Horseradish peroxidase-conjugated goat anti-mouse IgG (H + L) was used as the secondary antibody. The dilution ratios for the antibodies anti-GPX4, anti-DNMT3b, anti-FcRn, and anti-p53, as well as the companies they were sourced from, are consistent with those we previously described [28].
Quantitative real-time polymerase chain reaction
Total RNA was extracted from the lung tissue samples using TRIzol reagent (G3013-100; Servicebio, China). The quality and purity of the RNA were assessed by measuring the absorbance at 260–280 nm via a spectrophotometer. RNA was reverse transcribed into cDNA using an RT kit (MR0502-1; Kermey, China). SYBR Green PCR Master Mix (G3326-01; Servicebio, China) was used for real-time quantitative RT-qPCR to measure gene expression. Primers were synthesized by Suzhou Jun Among Biotechnology. Gene expression was normalized via the 2−ΔΔCT method, with the β-actin gene used as an internal standard. Each biological replicate consisted of three technical replicates (Table 2).
Table 2 Primers for RT-qPCRMDA detection
Lipid peroxidation was assessed using a malondialdehyde (MDA) measurement kit. This kit enables the measurement of MDA by reacting it with thiobarbituric acid (TBA), resulting in the production of a colorimetric reaction product with an absorption peak at 532 nm. The intensity of the color directly corresponds to the concentration of MDA. The MDA content was measured using the method we previously described [28].
Iron assay
The Fe2+ content was measured using the method previously described [25]. The reaction with the reagent was carried out by adding 20 μL of sample, standard, or distilled water to a 96-well plate labeled A test, A standard, or A blank, respectively. Absorbance was measured at 510 nm, and a standard curve was obtained at different concentrations. Fe2+ levels within the cells were calculated according to the manufacturer's instructions.
Analysis of glutathione (GSH) and glutathione disulfide (GSSG)
Intracellular GSH and GSSG levels were determined via GSH (BC1175; Solarbio, China) and GSSG (BC1185; Solarbio) quantification kits, respectively. The cells were repeatedly frozen, thawed, and sonicated (power 200 W, sonication 3 s, interval 10 s, repeated 30 times), and the resulting supernatant was centrifuged. The obtained sample was added to a 96-well plate and the absorbance was measured at 412 nm. A standard curve was created using different concentrations of GSH, and the GSH and GSH/GSSG contents within the cells were calculated via the provided formula.
Dual-luciferase reporter gene assay and ChIP
The TESS system was used to identify the c-Jun binding site in the FcRn promoter region. We elucidated the regulatory mechanisms underlying FcRn. For luciferase reporter assays, we constructed plasmids with distinct segments of the FcRn promoter region (F1 through F9), utilizing a range of primers (Table 3). Truncated promoter fragments were generated by PCR amplification. A549 cells were co-transfected with 0.2 μg of the luciferase reporter construct and 0.1 μg of the pRL-TK vector. After 24 h transfection, cells were exposed to HSV-1 at a multiplicity of infection (MOI) of 1, and luciferase activity was measured using the Promega Dual Luciferase Assay Kit following the manufacturer's protocol.
Table 3 Primers used for amplification and cloning of the FcRn gene promoterThe presence of a c-Jun binding site in the FcRn promoter was confirmed via ChIP assays. A549 cells were treated with HSV-1, fixed with 1% formaldehyde, and the nuclei were extracted. DNA was cleaved by sonication and ChIP was performed using an anti-DNMT3b antibody (with normal IgG as a negative control). DNA samples were subsequently amplified via specific PCR primers (Table 4).
Table 4 Primers used in the ChIP assaysStatistical analysis
The data were analyzed as the means ± SEMs, and all the data are expressed as the means ± standard deviation. Group comparisons were conducted using one-way analysis of variance (ANOVA), and statistical significance was assessed via unpaired t-tests or one- or two-way ANOVA. The software used for the analysis was GraphPad Prism 8.3.0 (GraphPad Software, San Diego, CA, USA). Significance was determined at levels of P < 0.05 ( ∗), P < 0.01 (∗ ∗), and P < 0.001 (∗ ∗ ∗), with a P-value of < 0.05 considered statistically significant.



留言 (0)