
記住我
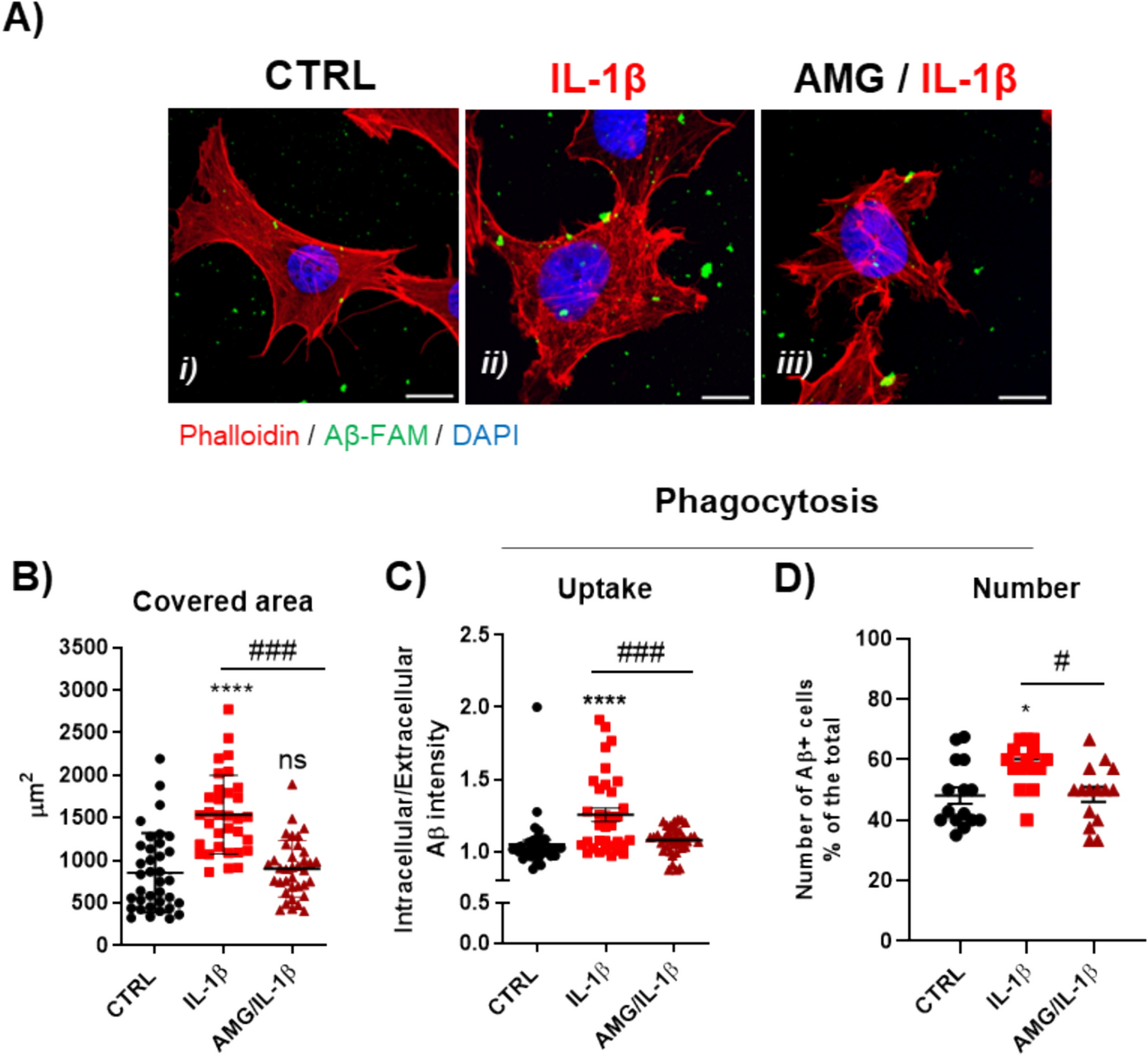


C20 cells, an established model of human microglia cells [40], were challenged with IL-1β, a well-known inducer of inflammatory response in the same cell line [39,40,41], and by using experimental conditions previously set [42]. To evaluate the effects of de novo neurosteroidogenesis on microglial reactivity, C20 cells were treated with AMG, the inhibitor of the first neurosteroidogenic enzyme CYP11A1 [42, 43], prior to IL-1β activation (AMG/IL-1β). AMG was shown to inhibit pregnenolone production in a dose-dependent manner over time, suggesting the specificity of the observed result. The 50 µM AMG concentration was chosen for the subsequent experiments, after ascertaining that it did not influence the viability of the C20 cells (Figures S1E). After treatments, morphological analysis and phagocytic activity, reported both as quantity of engulfed Aβ (uptake) and % of Aβ + microglia (number) were assessed (Fig. 1A). Control cells (CTRL) showed well-defined boundaries and branched morphology, typical of surveillant/homeostatic microglia phenotype (i). In contrast, IL-1β-treated microglia exhibited retractions of cell protrusions and an oval and spiny convexity shape, a phenotype named ameboid and commonly associated with activated states of microglia (ii) [44, 45]. Notably, the ameboid morphology of IL-1β-treated C20 cells was also associated to a highly significant enlargement of the cell body, measured as area covered by the cells (Fig. 1B), and higher propensity of the resulting microglia to β-amyloid peptide engulfment compared to control (Fig. 1C-D). On the other hand, AMG/IL-1β-treated microglia showed undelimited cell boundaries, unstructured cortical actin filaments, without a clear morphology (amoeboid or branched), resembling a dystrophic morphology (iii). Concomitantly, AMG/IL-1β microglia showed neither a significant increase in the area covered by the cell, nor in the phagocytic activity, compared to CTRL cells; however, both parameters were significantly impaired in comparison with IL-1β-challenged microglia, reducing the uptake and number of phagocytosing cells. In conclusion, AMG resulted to prevent human microglia from acquiring an activated and functional phenotype induced by the inflammatory stimulus, suggesting an intrinsic regulation of microglial activation processes by de novo produced neurosteroids.
Fig. 1
Characterization of microglia cell morphology and phagocytic activity in activated human microglia. A Representative confocal microscopy images of CTRL, IL-1β and AMG/IL-1β -treated C20 cells during phagocytosis assay displaying three merged channels: Aβ-FAM (green); Phalloidin-594 (red); DAPI staining (blue). Scale bar 10 μm. Confocal analysis were performed to evaluate cell morphology (B) and phagocytosis (C, D). In particular, a ROI was drawn for each cell and the area (μm2) of the red channel and fluorescence intensity of the green channel were measured by using ImageJ software. For the phagocytic activity, the human FAM-labelled β-amyloid peptide was used and the results are reported as ratio between intracellular and extracellular β-amyloid fluorescence intensity. A total of ten cells were analysed for each treatment in three independent experiment. The number of phagocytosing microglia was reported as % of the FAM-Aβ + cells. A number of 5 image fields for each treatment were analysed for three independent experiments. Data are represented as mean ± SEM of the pooled experiments and statistical analysis was determined by one-way ANOVA followed by Bonferroni's post-test: **** p < 0.0001, * p < 0.05 vs CTRL; ### p < 0.001 # p < 0.05 vs AMG/IL-1β C20 cells
To assess the dynamic regulation of the balance between pro- and anti-inflammatory microglia activation, experiments were performed at different time points after IL-1β treatment. We observed that in our model the treatment with AMG alone did not significantly influence the mRNA expression of pro- and anti-inflammatory cytokines (Figure S1B), therefore the attention was mainly directed towards the analysis of the inflammatory response following IL-1β treatment. Firstly, the nuclear translocation of the transcription factor NF-κB, which primarily occurs in response to the activation of IL-1β receptor [46, 47], was assessed by measuring the levels of p65, a protein being part of the NF-κB complex, in nuclear (N) and cytosolic (C) fractions (Fig. 2A; Figure S2A-B), after 4 and 24 h. Since nuclear p65 levels depend on both the translocation process and expression levels, which could be influenced by the inflammatory conditions themselves, the results were reported as ratio between nuclear or cytosolic p65, and the total p65 levels, as previously described [48]. C20 cells quickly respond to inflammatory stimulus, as indicated by increased p65 nuclear levels after 4 h of IL-β treatment (p = 0,0032 vs CTRL 4 h), whose levels tended to significantly reduce after 24 h of IL-β treatment (p = 0,028 vs IL-1β 4 h) (Fig. 2B). On the other hand, in AMG/IL-1β-treated cells, nuclear p65 levels were significantly increased after 24 h (p ≤ 0,0001 vs CTRL 24 h; p = 0,0012 vs IL-1β 24 h), suggesting a prolonged state of inflammatory pathway activation. In all conditions and time points, there were no significant differences in cytosolic and total p65 levels (Figure S1 C and D), implying, however, that in our model both increased nuclear translocation and total expression of p65 are necessary to maintain an activated inflammatory state. To further confirm the results of p65, an immunostaining of p65 was conducted to assess nuclear translocation. Firstly, cells were classified into “No Translocation” or “Translocation” depending on the localization area of p65 (cytosol or nuclear area; Figure S2C). In IL-1β treated cells, the percentage of cells with translocated p65 significantly increased after 4 h; however, the percentage of “No translocation” cells significantly increased at 24 h (p = 0,0059 vs IL-1β 4 h), further corroborating the activation of a controlled microglia inflammatory response. Notably, particular differences were observed between IL-1β and AMG/IL-1β groups in the percentage of cells in the “Translocation” state following 24 h of IL-1β (IL-1β: p = 0,0019 vs CTRL 24 h; AMG/IL-1β: p ≤ 0,0001 vs CTRL 24 h, p = 0,0021 vs IL-1β 24 h), suggesting a prolonged state of inflammatory activation and the establishment of a slowly recovering microglial phenotype following neurosteroidogenesis inhibition.
Fig. 2
AMG-mediated neurosteroidogenesis inhibition: analysis of pro- and anti-inflammatory markers in IL-1β-treated C20 cells. A Western blot analysis of nuclear (N) and cytosolic (C) fraction of p65 in CTRL, IL-1β and AMG/IL-1β-treated C20 cells after 4- and 24- hours from inflammation. Optimisation of the separation process of the N and C fractions was performed by assessing the presence of the H3 and GAPDH proteins. B Nuclear p65 densitometric analysis quantification normalized with respect to the total protein (Figure S1A). Data are reported as mean ± SEM of the proportion of Nuclear p65 (p65(N)/p65(Total)) and normalized against CTRL 4 h, obtained in three independent experiments. C-D Evaluation of p65 translocation by immunostaining analysis; bars are represented as percentages of cells with or without translocated p65. Scale bar 10 µm. E-J Quantitative RT-PCR of pro-(IL-6; TNF-a; NOX2) and anti-(IL-10; TGF-β; Arginase-1) inflammatory mediator/marker transcripts at different times of inflammation. Results are reported as mean ± SEM of the fold of change vs CTRL 4 h of three independent experiments. K) BDNF quantification in the MCM by ELISA assay. pg/mL values reported are normalized on absorbance values obtained from the crystal violet assay. Bars are represented as mean + SEM of three independent experiments. Statistical analysis was determined by two-way ANOVA followed by Bonferroni's post-test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs CTRL of each time point; °p < 0,05,°°p < 0,01 vs IL-1β of each time point; §p < 0.05, §§p < 0.01, §§§p < 0.001, §§§§p < 0,0001 vs previous time point
The microglial activated state was further evaluated by measuring the gene expression of proteins known to be involved in the pro- and anti-inflammatory response after 4, 24 and 36 h from IL-1β challenge (Fig. 2E-J). Following 4 h of IL-1β treatment, C20 cells increased gene expression of the cytokines IL-6 and TNFα (p = 0,041 and p = 0,031 vs CTRL 4 h) (Fig. 2E-F), as well as the expression of the key enzyme NOX2 involved in driving microglia ROS production (p = 0,005 vs CTRL 4 h) (Fig. 2G) [49, 50]. Notably, IL-1β-treated C20 cells quickly upregulated also the expression of IL-10 and Arginase-1 (p = 0,011 and p = 0,0034 CTRL 4 h) (Fig. 2H-I), commonly associated with a microglial restorative phenotype. Following 24 h of IL-1β treatment, the expression of most of the investigated genes significantly decreased, although major reductions were observed for IL-10 and Arginase-1 (IL-1β 24 h: IL-10 p = 0,0001; Arginase-1 p < 0,0001 vs IL-1β 4 h), whereas the expression of IL-6 and TNF-α still remained higher than in CTRL C20 cells. On the other hand, TGF-β gene expression was found to be upregulated at 24 h of inflammation (IL-1β 24 h: TGF-β p = 0,036 vs IL-1β 4 h) (Fig. 2J). At 36 h, only the expression of IL-6 again significantly increased (IL-1β 36 h: IL-6 p = 0,034 vs CTRL 24 h, p < 0,0001 vs IL-1β 24 h), suggesting a key role for this cytokine in the maintenance of the inflammatory response. Notably, the expression of TGF-β was further increased at 36 h (p = 0,036 vs CTRL 36 h), suggesting a key role for this cytokine in promoting controlled microglial activities, consistent with TGF-β homeostatic function in microglia [51]. Taken together, these findings indicate that when exposed to the inflammatory IL-1β stimulus, C20 cells polarized and properly mounted a controlled inflammatory response, a process that is essential for microglia to effectively perform their main effector functions. Notably, a different scenario was observed when neurosteroidogenesis was inhibited (AMG/IL-1β). Firstly, AMG pretreatment appeared to initially inhibit the inflammatory response to IL-1β, as no significant changes in gene expression of both pro- and anti-inflammatory associated proteins were observed after 4 h, except for NOX2 (p = 0,079 vs CTRL 4 h). After 24 h, however, only a pro-inflammatory response was observed, characterized in particular by a strong increase in TNF-α and NOX2 expression (p = 0,0060 and p = 0,0014 vs AMG/IL-1β 4 h), indicating an imbalance in the profile of microglial pro- and anti-inflammatory activities. Notably, TGF-β gene expression was downregulated also after 24 h of inflammation. Overall, after 24 h, the results might suggest a delay due to AMG pre-treatment in the activation of a correct inflammatory response. However, following 36 h of IL-1β treatment, we observed a drastic increase in IL-6 expression, a slight reduction of TNF-α, and a significant reduction of NOX2 (p = 0,0026 vs AMG/IL-1β 24 h), without changes in the expression of IL-10 and Arginase-1, whose levels still maintained downregulated. Moreover, TGF-β expression levels increased with respect to 24 h. Overall, these results reinforce the idea that AMG compromises the balance of pro- and anti-inflammatory responses; however, the increasing trend in TGF-β gene expression could represent the attempt of AMG/IL-1β cells, albeit delayed, to restore a controlled response.
Finally, the neurotrophic function of microglia was assessed by analyzing BDNF levels released in the media after 24 h treatment (Fig. 2I). As expected, inflammation significantly reduced BDNF levels; however, inhibition of neurosteroidogenesis by pre-treatment with AMG further impaired BDNF release, suggesting the complete loss of neurotrophic activities by microglia [52]. Overall, the data from AMG-treated C20 cells indicated that the inhibition of neurosteroidogenesis impairs microglial functional reactivity, presumably resulting in a hyper-activated inflammatory state.
Neurosteroidogenesis inhibition suppressed the homeostatic TGF-β pathway in human activated microgliaTGF-β signalling is known to be a key regulator for the maintenance of a surveillant microglia phenotype [52, 53] and results to be suppressed in disease-associated and aged microglia [9], thus hindering the restoration of a homeostatic microglial state. This signalling is activated when TGF-β interacts with the type 2 receptor (TGFβR2), which phosphorylates the type 1 TGF-β receptor (TGFβR1) that in turn phosphorylates its primary targets, Smad2 and Smad3. Phosphorylated Smad2/3 complex translocates into the nucleus as heterodimer with Smad4, promoting the transcription of targeted genes, as MMP-2, MMP-9 and Integrin β5 that are in turn involved in the trans-activation of TGF-β [51].
Given the prolonged inflammatory state and the unbalanced inflammatory response in AMG/IL-1β-treated C20 cells, including the downregulation in gene expression of TGF-β, the TGF-β signalling pathway was kinetically monitored by measuring the gene expression of the effector proteins involved in the signalling (Fig. 3A-D), as well as TGF-β targeted genes (Fig. 3E–G). In IL-1β group, the expression of the first effector of the signalling, TGFBR2, followed that of its endogenous ligand TGF-β, with an initial reduction at 4 h and a significant recovery after 24 h (p = 0,0011 vs IL-1β 4 h). Furthermore, TGFBR1 was significantly increased too. In contrast, Smad2 and Smad4 levels were increased at 4 h, and decreased over time to control levels. The expression of all TGF-β signalling target genes, MMP2, MMP9 and Integrin-β5, increased following 24 h of inflammation. Overall, the results suggested an initial downregulation of signalling probably due to reduced TGFBR2 expression (the primary receptor driving the pathway activation in response to exogenous TGF-β), and at the same time its maintenance by increased Smad2/4 expression. Later points confirmed that the IL-1β induces an efficient activation of the TGF-β pathway in C20 cells, leading to a sustained up-regulation of target genes.
Fig. 3
Evaluation of the effects of neurosteroidogenesis inhibition on TGF-β signalling in activated C20 cells. A-G mRNA expression levels of proteins promoting the TGF-β signalling pathway (TGFBR2/ TGFBR1/Smad2/Smad4) and target genes (MMP2/MMP9/Integrin β5) by quantitative Real-Time RT PCR following 4, 24 and 36 h from IL-β induction. Results are reported as mean ± SEM of the fold of change vs CTRL 4 h of three independent experiments. Statistical analysis was determined by two-way ANOVA followed by Bonferroni's post-test: *p < 0.05, **p < 0.01, ****p < 0.0001 vs CTRL of each time point; °p < 0,05,°°p < 0,01 vs IL-1β of each time point; §p < 0.05, §§§§p < 0,0001 vs previous time point. H) Western blot analysis of phosphorylated Smad2 and Smad3 (p-Smad2/p-Smad3) in CTRL, IL-1β-treated and AMG/IL-1β-treated C20 cells after the exposition to TGF-β (20 ng/ml) for 4 h. I) Densitometric analysis of bands concerning p-Smad2/p-Smad3, quantified with respect to the total protein (Figure S2 B). Bars are represented as mean ± SEM and statistical analyses were determined by one-way ANOVA in each group (w/o TGF-β or with TGF-β) followed by Bonferroni’s post-test: **p < 0,01 vs CTRL; ° p < 0,05 vs IL-1β
On the other hand, in AMG/IL-1β-treated sample, the activation of the signalling appears to be impaired, as suggested by the maintained downregulation of TGFBR2 at every investigated time points. Moreover, the downregulation was not supported by the Smad proteins as they resulted downregulated after 4 h of inflammation compared to IL-1β group (Smad2: p = 0,0002; Smad4: p = 0,0014 vs IL-1β). However, a recovery, in the case of MMP-2, and an up-regulation of MMP9 and integrin β5 was observed after 36 h. This result supports the hypothesis that AMG might delays the ability of microglia to keep the control over the inflammatory response. Indeed, AMG did not negatively impact on TGFBR1 gene expression, suggesting the potential contribution of this receptor in the observed activities. However, verification of this hypothesis and investigation of the precise alternative signalling through this receptor require further studies.
Moreover, human microglia were treated with exogenous TGF-β, and the amount of phosphorylated heterodimer Smad2/Smad3 (p-Smad2/3) was investigated to evaluate the propensity of the system to restore homeostatic conditions (Fig. 3H-I). As expected, exogenous TGF-β treatment highly increased p-Smad2/3 levels in C20 CTRL cells, indicating an effective activation of the TGF-β signalling in surveillant microglia. Notably, inflammation per se induced the activation of the TGF-β pathway as indicated by increased p-Smad2/3 levels in C20 IL-1β, suggesting that inflamed microglia basally stimulate TGF-β signalling to avoid their overactivation. C20 IL-1β cells were still responsive to TGF-β stimulus, although a lower increasing in p-Smad2/3 was observed compared to C20 CTRL cells. On the other hand, a significant increase in p-Smad2/3 levels was not observed in AMG/IL-1β w/o TGF-β, whereas in the presence of TGF-β, the response was lower compared to CTRL and IL-1β (p = 0,007 vs CTRL; p = 0,026 vs IL-1β), implying an overall less responsive system to the homeostatic/restorative signal.
In line with the increased expression of TGF-β following 24 h of inflammation, these findings suggest that TGF-β signalling plays an important role in the regulation of controlled activation of microglia. Microglia resulting from the inhibition of neurosteroidogenesis showed suppression of the TGF-β signalling, corroborating the idea of the establishment of a chronically activated and dysfunctional microglia phenotype.
TSPO ligand-mediated de novo neurosteroidogenesis stimulation promotes restorative phenotype in human microgliaTo further investigate the role of neurosteroidogenesis on microglia phenotype, TSPO was pharmacologically stimulated to enhance de novo neurosteroidogenesis. Thus, C20 cells were pre-treated with 1 μM of the highly steroidogenic TSPO ligand (XBD-173 or PIGA1138) [13] for 2 h, and then exposed to IL-1β for 22 h. Morphological analyses evidenced a more polarized/bushy phenotype in TSPO ligand pre-treated C20 cells. Moreover, the pre-treatments slightly reduced the area covered by the cells and the Aβ-FAM uptake (IL-1β: p = 0,0007; XBD-173/IL-1β: p = 0,013; PIGA1138/IL-1β: p = 0,022 vs CTRL), however, both parameters did not significantly differ compared to C20 IL-1β (Fig. 4B-C). Conversely, an increased trend in the number of phagocytosing microglia was observed in TSPO ligands pretreated cells (IL-1β: p = 0,0018; XBD-173/IL-1β: p = 0,0004; PIGA1138/IL-1β: p = 0,0002 vs CTRL) (Fig. 4D).
Fig. 4
TSPO-mediated neurosteroidogenesis stimulation: Analysis of pro- and anti-inflammatory markers in IL-1β-treated C20 cells. C20 cells were pre-treated with 1 μM of the selective TSPO ligand XBD-173 or PIGA1138 for 2 h; then the inflammatory stimulus was administered. A Representative confocal microscopy images of Aβ-FAM (green) and Phalloid-594 (red) of CTRL, IL-1β, PIGA1138/IL-1β and XBD-173/IL-1β-treated C20 cells. DAPI staining is used to mark cell nuclei (scale bar 10 μm). Confocal analyses were performed to evaluate cell morphology (B) and phagocytosis (C-D) following 24 h of inflammation. A total of ten cells were analysed for each treatment in three independent experiments. For the number of phagocytosing microglia, 5 image fields for each treatment were analysed for three independent experiments. Data are represented as mean ± SEM of the pooled experiments and statistical analysis was determined by one-way ANOVA followed by Bonferroni's post-test: *p < 0,05, **p < 0,01, ***p < 0,001, ****p < 0.0001 vs CTRL. E-J Real-time RT PCR analyses were performed for the mRNA expression level quantification of pro- and anti-inflammatory genes at different time points from IL-1β treatment. Results are reported as mean ± SEM of the fold of change vs CTRL 4 h of three independent experiments. Statistical analysis was determined by two-way ANOVA followed by Bonferroni's post-test: * p < 0.05, **** p < 0.0001 vs CTRL of each time point; °p < 0,05,°°p < 0,01, °°°p < 0,001, °°°°p < 0,0001 vs IL-1β of each time point; §§p < 0.01, §§§p < 0,001, §§§§p < 0,0001 vs previous time point. K) The microglial conditioned medium was collected after 24 h of treatment and analysed for its BDNF content by ELISA assay. Bars are represented as means ± SEMs of three independent experiments and statistical analyses were determined by one-way ANOVA followed by Bonferroni's post-test: **p < 0.01 vs CTRL; °p < 0,05 vs IL-1β
Regarding the inflammatory response (Fig. 4E-J), conversely to AMG inhibition, both TSPO ligands were able to attenuate the expression of proteins classically involved in the pro-inflammatory response, and to elicit anti-inflammatory ones. TSPO ligands reduced the expression of IL-6 and TNF-α induced by IL-1β at every time points and induced the anti-inflammatory response by increasing the expression of IL-10 and Arginase-1, in particular following 36 h from IL-1β treatment. Interestingly, these effects were markedly evident in sample pre-treated with the highest steroidogenic ligand PIGA1138, and furthermore, only PIGA1138 was able to induce a significant expression of TGF-β starting from 24 h of inflammation (PIGA1138/IL-1β 24 h: p = 0,0040 vs CTRL 24 h, p = 0,0034 vs IL-1β 24 h; PIGA1138/IL-1β 36 h: p = 0,0002 vs CTRL 36 h, p = 0,043 vs IL-1β 36 h). However, an increased expression of NOX2 could be observed at 36 h following treatment with TSPO ligands compared to IL-1β, suggesting either a possible increase in mitochondrial metabolism or a delay in the upregulation of NOX2 gene expression compared to C20 IL-1β. Finally, XBD-173 and PIGA1138 were able to restore BDNF levels (analysed at 24 h), although significant levels were observed only for PIGA1138 (p = 0,034 vs IL-1β).
Taken together, these results suggest that TSPO-mediated enhancement of neurosteroidogenesis mitigates the pro-inflammatory response induced by the immunogenic stimulus, promoting instead an activated microglial phenotype that maintains phagocytic activities and simultaneously attempts to acquire an anti-inflammatory and reparative phenotype.
3.4 Neurosteroidogenesis inhibition and stimulation differently influenced the expression of genes associated to homeostatic and functional/dysfunctional-associated microglial statesIn order to further characterize the microglial phenotype resulting from the inhibition or stimulation of neurosteroidogenesis, the expression of specific genes associated with homeostatic or functional/ dysfunctional microglial states was investigated in C20 cells at 36 h after the inflammatory stimulus. In particular, genes associated with immunoregulation (CX3CR1; Figs. 5A) [54], surveillance activities (P2RY12; Figs. 5B) [55], microglial signature (HexB; Fig. 5C) [56], lipid homeostasis (ApoE; Figs. 5D) [57], phagocytosis (Trem2, Tyrobp; Figs. 5 E–F) [54], and primed state [11] (CD86; HLA-DRA; Figs. 5 G-H) were taken into account. IL-1β treatment induced an activated phenotype with increased surveillance and phagocytic capacities, consistent with the establishment of a controlled activated phenotype. In contrast, pre-treatment with AMG induced a senescent-like and primed phenotype characterized by a complete loss of the homeostatic and microglial signature, reduced expression of phagocytosis effector proteins, and a drastic increase in the expression of proteins involved in the establishment of chronic inflammatory state. Furthermore, the results also suggested a deregulated state of lipid homeostasis, as indicated by increased expression levels of ApoE. Interestingly, pre-treatment with TSPO ligands further induced the expression CX3CR1, P2RY12, Trem2, and Tyrobp compared to IL-1β group, suggesting the achievement of a regulatory and reparative microglial phenotype. Once again, major effects were observed following PIGA1138 treatment. Overall, the results further emphasized that regulation of endogenous neurosteroid production in microglia plays a key role in maintaining a functionally activated phenotype.
Fig. 5
Gene expression analysis of known markers associated with homeostatic and dysfunctional microglial phenotypes following inhibition or stimulation of endogenous neurosteroid production. Real-Time RT-PCR analysis of genes associated with homeostasis and surveillant activities (A, B), lipid homeostasis (C-D), phagocytosis (E, F) and propagation of the inflammatory state (G, H). Bars are represented as means ± SEMs of three independent experiments and statistical analyses were determined by one-way ANOVA followed by Bonferroni's post-test: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 vs CTRL; #p < 0.05; ##p < 0.01; ###p < 0.001; ####p < 0.0001 vs IL-1β
3.5 The microglial conditioned medium resulting from the inhibition or TSPO-mediated stimulation of de novo steroidogenesis differently impacts on the proper differentiation of human neural progenitor cellsThe analysis conducted so far has allowed us to deeply characterise the phenotypic shifting induced by the inhibition or stimulation of de novo neurosteroidogenesis. However, whether this can lead to microglial states with beneficial or detrimental functions may remain speculative (e.g. IL-6 could be pro- and anti-inflammatory at the same time, depending on the context [58]). To this end, a functional proof was conducted by evaluating the effects of the resulting MCMs on the activities and differentiation of hNPCs (Fig. 6A). Microglia present a high capacity to recall neural stem cells (NSCs) to the site of injury through their secretome and to promote their differentiation into mature CNS cells [59, 60]. However, depending on the degree of microglia activation, the secretome can negatively affect NSC pool viability and differentiation fate. To this end, MCMs were collected from treated C20 cells [CTRL; IL-1β; AMG/IL-1β; XBD-173/IL-1β; PIGA1138/IL-1β] and added to hNPCs; then cell viability, migratory activity and differentiation fate were evaluated (Fig. 6A). As already reported [59], the concentration of the MCM was set at 10% in order to avoid any effects related to the change in the type of medium. As control samples (Untreated), hNPCs were treated with 10% of non-conditioned C20 culture medium.
Fig. 6
Different microglial activation profiles influence human NPCs activities and differentiation fate. A MCM derived from CTRL, IL-1β, AMG/IL-1β, XBD-173/IL-1β, PIGA1138/IL-1β C20 cells were used as treatment to evaluate the effects of different microglia activation profiles on the hNPCs activity and differentiation process. B Cell viability of hNPCs treated with the four different MCMs was evaluated by MTS assay. The results were expressed in percentage (%) vs untreated hNPCs. C Migratory potential of hNPCs was assessed by the Boyden chamber assay, in which the lower part of the well was filled with complete culture medium alone or in combination with 10% of FBS, used as positive control, or the four different MCMs. The results were analysed by manual count of migrated cells. C Representative microscopy images of untreated hNPCs and treated with the four different MCMs for Nestin (green), GFAP (red) and DAPI (blue) at left, and for Tuj1 (green), GFAP (red) and DAPI (blue) at right (scale bar 20 μm). E Fluorescence intensity analysis of Nestin in differentiated hNPCs. On the same images, the number of Nestin-/GFAP + cells were analysed (F). G Neuronal commitment of hNPC was quantified by the ratio between the number of Tuj1 + cells and the total number of cells. H Neuronal development was assessed by neurite density values obtained by calculating the ratio between the total length of neurites and the total number of Tuj1 + cells. Bars are represented as mean + SEM of three independent experiments and statistical analysis was determined by one-way ANOVA followed by Bonferroni's post-test: *p < 0.05; **p < 0.01; ****p < 0.0001 vs Untreated NPCs; #p < 0.05; ##p < 0.01; ####p < 0.0001 vs MCM CTRL; °p < 0,05 vs MCM IL-1β
Firstly, cell viability of undifferentiated hNPCs was evaluated by MTS assay following the addition of 10% of the four different MCMs to hNPCs complete culture medium (Fig. 6B). After 72 h, none of the MCMs significantly influenced the viability of hNPCs. As another key function of hNPCs, their migratory potential was assessed by the Boyden chamber assay (Fig. 6C). The lower part of the well was filled with complete culture medium alone or in combination with 10% of FBS, used as positive control, or the four different MCMs. Results demonstrated that exposure to MCMs significantly increased the migratory activity of hNPCs with levels comparable to FBS, suggesting an intrinsic chemoattractant effect of the MCM independently from the microglia polarization state. Although significant differences between MCMs were not observed, C20 IL-β and AMG/IL-1β derived MCM slightly reduced the chemoattractant capacity of C20 CTRL cells, whereas TSPO ligand pre-treatment counteracted this effect.
Differentiation of hNPCs can be induced by mitogen factors withdrawal. Several evidences report that in the early stage the NPCs resulted Nestinhigh/GFAP+; after the induction of the proliferative phase and differentiation processes, cells lose Nestin expression with the commitment of hNPCs to glial- or neural-fate [61]. Herein, hNPCs were exposed to 10% of the various types of MCMs after growth factors withdrawal and let them to differentiate for 6 days, chosen as the stage at which neuronal and astrocytic markers reached the maximum expression levels (Figure S5A). Cells were stained for Nestin as a marker of proliferating and still undifferentiated cells, Tuj1 (βIII-Tubulin) and GFAP as neuronal and astrocytic markers, respectively (Fig. 6D). Immunocytochemistry experiments were analysed for the fluorescence intensity of Nestin signal and for the number of Nestin−/GFAP+ cells, as indexes of the speed of NPCs differentiation (Fig. 6E-F); for the number of Tuj1+ cells (Ratio #Tuj1+/ Total # of cells) and neurites density (μm neurites/ # of Tuj1+ cells; Figure S5B) (Fig. 6G-H) to characterize the neurogenic ability of hNPCs under the influence of MCM. In the presence of all MCMs, Nestin expression was significantly reduced, suggesting that the presence of soluble factors released by microglia could promote the proliferation and differentiation of hNPCs. However, most cells treated with MCM CTRL maintained a polarised morphology, and furthermore, the presence of MCM CTRL promoted neurite outgrowth, suggesting that microglia under homeostatic conditions may contribute to establish a microenvironment capable of providing neurotrophic support to hNPCs. Conversely, the morphology of cells treated with MCM IL-1β more closely resembled that of a mature astrocyte. Indeed, under these conditions, Nestin expression was further reduced (p = 0,0011 vs MCM CTRL), and most cells were Nestin−/GFAP+(p = 0,007 vs Untreated; p = 0,027 vs MCM CTRL). Also, the proportion of neural-committed cells and neurites density were also highly reduced following treatment with MCM IL-1β, further underlining how the shift in the balance towards a pro-inflammatory microglial phenotype is more likely to favour differentiation towards an astrocytic population. This aspect was further confirmed by the results obtained with MCM derived from microglia pre-treated with AMG or TSPO ligands. Notably, the detrimental effects of MCM IL-1β were exacerbated by the pre-treatment with AMG, as suggested by dramatic decrease in Nestin expression and neuronal differentiation (p = 0,036 vs MCM IL-1β), further corroborating the establishment of a dysregulated microglial phenotype. On the other hand, the anti-inflammatory microglial phenotypic shift was further confirmed as hNPCs treated with MCM XBD-173/IL-1β or PIGA1138/IL-1β presented restored Nestin levels and reduced number of Nestin-/GFAP + cells. Notably, both MCMs derived from pre-treated cells with TSPO ligands resulted in a rescue of the neuronal-committed cells and neurites density; although only the MCM PIGA1138/IL-1β led to a significant restoration of the neuritic density compared to MCM IL-1β (p = 0.017 vs MCM IL-1β).
Overall, the results showed that stimulation of endogenous microglial neurosteroidogenesis by TSPO ligands was able to effectively promote a reparative phenotype that could counterbalance the damaging effects induced by pro-inflammatory conditions on the differentiation process of hNPCs. Importantly, as little as 10% of MCM was able to induce significant changes, and as the secretome contains several types of molecules, including neurosteroids, the results confirmed that the latter may indeed play a key role in driving hNPC differentiation.
3.6 Peculiar neurosteroidome profiles drive human microglia towards the acquisition of pro-inflammatory and reparative phenotypesResults obtained suggested a possible key role for de novo neurosteroidogenesis (summarized in Fig. 7) in regulating different functional microglial phenotypes.
留言 (0)