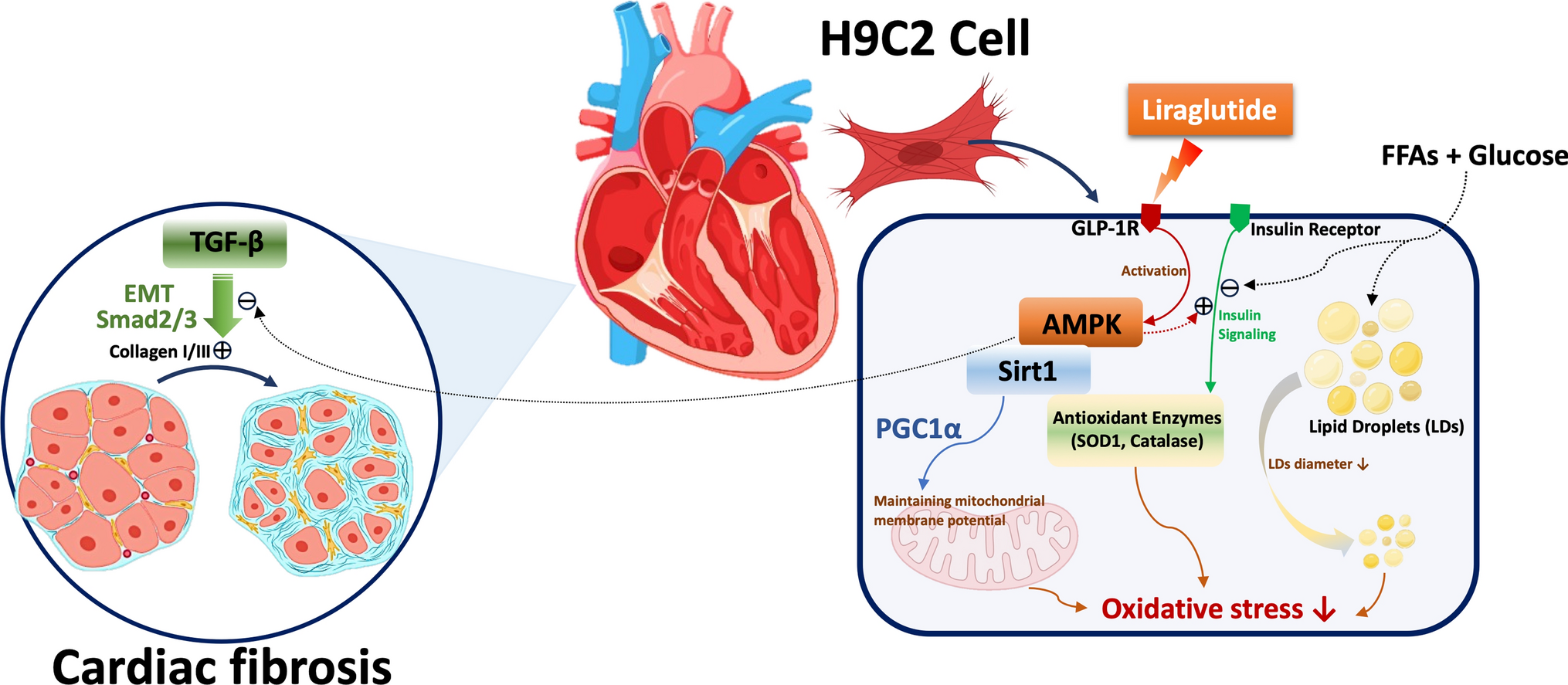
Materials
Sigma-Aldrich (Munich, Germany) supplied the chemicals used in this study, including 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 4′,6-diamidino-2-phenylindole (DAPI), 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), LY24002, compound C, JC-1, and Nile red. Antibodies were sourced from several suppliers: Santa Cruz Biotechnology (Santa Cruz, CA, USA) provided PGC1α (sc-517380; RRID:AB_2755043), Akt (sc-5298; RRID:AB_626658), phospho-Akt (sc-514032; RRID:AB_2861344), Talin 1 (sc-81805; RRID:AB_2303406), Integrin β1 (sc-374429; RRID:AB_11012020), and TGF-β1 (sc-130348; RRID:AB_1567351). GeneTex (Irvine, CA, USA) supplied antibodies for vimentin (GTX100619; RRID:AB_1952557), SOD1 (GTX100659; RRID:AB_1951972), catalase (GTX110704; RRID:AB_1949848), and Sirt1 (GTX61042; RRID:AB_10619663). ABclonal (Woburn, MA, USA) provided antibodies for GLP-1R (A13990; RRID:AB_2760844), αSMA (A2235; RRID:AB_2862980), and E-cadherin (A20798; RRID:AB_3107194), while Cell Signaling Technology (Danvers, MA, USA) supplied AMPK (#2532; RRID:AB_330331), phospho-AMPK (#2531; RRID:AB_330330), Smad2/3 (#5678; RRID:AB_10693547), and phospho-Smad2/3 (#8828; RRID:AB_2631089). Antibodies for IRS1 (05–1085; RRID:AB_1977296) and phospho-IRS1 (05–1087; RRID:AB_1977300) were obtained from Millipore (Bedford, MA, USA), and β-actin (NB600-501; RRID:AB_10077656) from Novus Biologicals (Littleton, CO, USA). Collagen I (ARG21965) and Collagen III (ARG20786) were acquired from Arigo Biolaboratories (Burlington, NC, USA), and liraglutide was sourced from Novo Nordisk (Copenhagen, Denmark). All chemicals were dissolved in phosphate-buffered saline (PBS) and stored at -20 °C until required for experimentation.
Cell culture and differentiation
Rat H9c2 cardiac myoblast cells (RRID:CVCL_0286) were obtained from the American Type Culture Collection (Bethesda, MD, USA). Cells were cultured in 75 cm2 tissue culture flasks using DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. Cultures were maintained at 37 °C with 5% CO₂ in a humidified atmosphere. To preserve differentiation potential, the supplier recommended subculturing the cells before reaching 70–80% confluence. When the cells reached approximately 50–60% confluence, the culture medium was replaced with differentiation medium. This differentiation medium, consisting of DMEM with 1% FBS and 10 nM all-trans retinoic acid (ATRA), was refreshed every 2–3 days, and the cells were maintained in this medium for 11 days. Differentiation was confirmed by observing morphological changes under a microscope, including cell elongation and alignment.
Immunocytochemistry
After treatment, ells were rinsed with PBS up to three times following removal of the culture medium. Cells were then fixed by adding 4% paraformaldehyde (PFA) in PBS to each well and incubated for 15–20 min at room temperature. Residual PFA was removed by washing the cells three times with PBS. To permeabilize, cells were incubated with 0.1% Triton X-100 in PBS at room temperature for 10 min. After three PBS washes, non-specific binding sites were blocked by incubating cells overnight at 4 °C in a humidified chamber with a blocking solution. Cells were then incubated in a humidified chamber at 4 °C overnight with the primary antibody diluted 1:100. Afterward, cells were incubated with the secondary antibody, diluted 1:200, in the dark at room temperature for 1 h. Stained cells were examined using the ImageXpress Micro Confocal High-Content Imaging System (Molecular Devices, Sunnyvale, CA, USA).
Nile red staining and high-content analysis (HCA)
Following treatment, cells were fixed with 4% paraformaldehyde for 15 min, then rinsed with PBS. Nile red staining (1 μg/mL) was performed in the dark at room temperature for five minutes. Imaging was carried out using the ImageXpress Micro Confocal High-Content Imaging System (Molecular Devices, Sunnyvale, CA, USA). Quantitative analysis of Nile red-stained particles was automated with a custom module in MetaXpress software (Molecular Devices, Sunnyvale, CA, USA), examining a minimum of 100 cells per well, with each condition performed in triplicate.
mRNA expression analysis by reverse-transcription quantitative PCR (qPCR)
Total RNA was isolated from cultured cells using Trizol reagent (Invitrogen, USA) according to the manufacturer’s instructions. After extraction, RNA purity and concentration were assessed by measuring absorbance at 260/280 nm with a spectrophotometer. The RNA was then reverse-transcribed into complementary DNA (cDNA) using a high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific, Waltham, MA, USA). Quantitative real-time PCR was performed using a Roche LightCycler 480 Real-Time PCR system (Roche, Basel, Switzerland) with SYBR Green PCR Master Mix (Roche, Basel, Switzerland) for mRNA detection. Reactions were conducted in a 20 μL volume containing 10 μL of SYBR Green Master Mix, 1 μL of each primer (10 μM), 2 μL of cDNA template, and 6 μL of nuclease-free water. GAPDH was used as the internal control for normalization. The thermal cycling conditions were set as follows: an initial denaturation at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s. Primer sequences are provided in Table 1. Melt curve analysis was conducted at the end of each run to confirm specificity of the amplification. Relative expression levels were calculated using the 2−ΔΔCT method, normalizing to GAPDH and comparing to control samples. All samples were run in triplicate to ensure data reliability, and negative controls (no-template controls) were included to rule out contamination.
Table 1 Primer sequence of different genes for qPCR analysisWestern blot analysis
Cells were washed twice with cold PBS and lysed using Gold lysis buffer (10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 0.5% sodium deoxycholate, 0.1% SDS, and protease/phosphatase inhibitors). For animal tissue samples, tissue was homogenized in T-PER Tissue Protein Extraction Reagent (Thermo Fisher Scientific, Waltham, MA, USA) at a ratio of 1:20 (w/v), tissue to T-PER reagent, to ensure efficient extraction. The homogenate was then centrifuged at 12,000 × g for 15 min at 4 °C to pellet cell or tissue debris. The supernatant containing the total protein extract was collected, and protein concentration was determined using a BCA protein assay kit (BioRad, Hercules, CA, USA), following the manufacturer’s protocol. Equal amounts of protein (20–40 µg) from each sample were loaded into each well of an SDS-PAGE gel. Following electrophoresis, proteins were transferred to a PVDF membrane using a wet transfer apparatus. Membranes were blocked with blocking buffer and then incubated with primary antibodies (1:1000) at 4 °C overnight with gentle agitation. After washing, membranes were incubated with secondary antibodies (1:5000) for 1 h at room temperature with gentle agitation. Signal detection was performed using Amersham ECL detection kits (Cytiva, Marlborough, MA, USA), and signals were visualized using an AI600 imaging system (GE Healthcare, Chicago, IL, USA). Western blot band quantification was conducted using Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA).
Measurement of reactive oxygen species (ROS)
Following treatment, cells were incubated with DCFH-DA (10 μM) for 30 min to allow ROS-dependent conversion of DCFH-DA to the fluorescent DCF. Cells were then washed and resuspended in PBS. Fluorescence intensity, indicative of ROS levels, was measured using the SpectraMax iD5 multi-mode microplate reader (Molecular Devices, Sunnyvale, CA, USA) with excitation at 485 nm and emission at 535 nm. For normalization, ROS levels in treated samples were quantified as a percentage relative to the untreated control. This was achieved by calculating the fluorescence intensity ratio between treated samples and controls. Specifically, untreated control samples were measured to establish baseline ROS fluorescence, and ROS levels in treated samples were calculated using the formula:
$$ } \left( \% \right) = \frac}}} \times 100 $$
Each treatment condition was assessed in triplicate to ensure reproducibility. This normalization allowed for direct comparison of ROS levels across different treatments, reported as a percentage of the control ROS level.
Lipid peroxidation measurement
Following treatment, cells were harvested by centrifugation at 3000 rpm for 10 min, and the supernatant was discarded. The cell pellets were resuspended in 300 µL of 10% medium and sonicated on ice for one minute. After adding 100 µL of precooled PBS, the cells were sonicated again and stored at -20 °C. Malondialdehyde (MDA) content, indicative of lipid peroxidation, was measured using the Lipid Peroxidation Assay Kit (BioVision, San Francisco, CA, USA) with colorimetric detection at 532 nm. Protein concentration was determined using a BCA protein assay kit (Millipore, Bedford, MA, USA), and MDA levels were normalized to protein content, expressed as nmol MDA per mg protein.
Mitochondrial membrane potential analysis
Mitochondrial functionality was evaluated using the cationic dye JC-1, which accumulates in mitochondria in a membrane potential-dependent manner. In healthy mitochondria, JC-1 forms red fluorescent aggregates, whereas in depolarized mitochondria under stress conditions, it remains as a monomer, emitting green fluorescence. After treatment, cells were incubated with 1 µM JC-1 at 37 °C for 30 min. Following incubation, the staining solution was removed, and cells were rinsed with PBS. Imaging was performed using an inverted fluorescence microscope (DP72/CKX41, Olympus, Tokyo, Japan). The red-to-green fluorescence ratio, indicative of mitochondrial membrane potential, was quantitatively analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Animal model preparation and experimental grouping
A schematic of the experimental design is shown in Fig. 5A. The study was conducted on 18 male db/db mice (Leprdb/db, 5 weeks old) and 12 male wild-type (WT) mice (C57BL/6, 5 weeks old). All animals were housed in a specific pathogen-free (SPF) facility with controlled temperature (18–22 °C) and humidity (30–70%). Mice were given ad libitum access to water and food. After one week of acclimatization on a normal chow diet (5% fat by weight), mice were randomly divided into five groups (n = 6) and assigned to the following dietary regimens for 24 weeks: normal chow, high-fat diet (HFD; 60% fat by weight, Envigo, Indianapolis, IN, USA), normal chow with C57BL/6 mice, HFD with C57BL/6 mice, normal chow with db/db mice, HFD with db/db mice, and HFD with db/db mice plus liraglutide. Body weight and fasting blood glucose levels (from tail vein samples) were recorded every four weeks. Liraglutide (0.4 mg/kg) was administered subcutaneously twice weekly to the designated groups for 24 weeks. At the study’s conclusion, mice were anesthetized and euthanized to collect blood and tissue samples. Hearts were preserved in 4% buffered formalin for subsequent analysis. This study was approved by the Institutional Animal Care and Use Committee (IACUC) of Chung Shan Medical University (Approval No. 2673).
Histological examination and immunofluorescence staining
Following euthanasia, heart tissue was harvested and fixed in 4% neutral buffered formalin for 24 h. For cryosectional histological analysis, the heart was embedded in an optimal cutting temperature (OCT) compound and rapidly frozen using dry ice or liquid nitrogen. Sections were prepared at 16 µm thickness using a cryostat (CM3050S, Leica Biosystems, Nussloch, Germany) and collected onto pre-labeled, frost-free glass slides. Fibrosis in the heart was visualized by staining sections with Picrosirius red. Immunofluorescence staining was conducted by incubating sections with primary antibodies, followed by fluorescently labeled secondary antibodies. Nuclei were counterstained with DAPI in the mounting medium. Images were acquired using a fluorescence microscope (BX53, Olympus, Tokyo, Japan).
Statistical analysis
Data are presented as means ± standard error of the mean (SEM) and were analyzed using analysis of variance (ANOVA) followed by Dunnett’s post-hoc test for multiple comparisons. Statistical analyses were performed using SPSS software (SPSS Inc., Chicago, IL, USA). A p-value of less than 0.05 was considered statistically significant.



留言 (0)