
記住我

All animal maintenance and experimental procedures were approved by the Stanford University Administrative Panel for Laboratory Animal Care and conformed to the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals. Efforts were made to minimize the number of mice used and their suffering. For all studies, mice were group-housed under a reverse light-dark cycle with lights off at 8:30 AM and on at 8:30 PM. Exceptions to group housing were made when individual mice were separated due to in-cage fighting. Food and water were freely available.
Chemogenetic LC inhibition (DREADD) studiesMice expressing Cre recombinase from the dopamine beta hydroxylase locus (DBH-cre; derived from B6.FVB(Cg)-Tg(dbh−Cre)KH212Gsat/Mmucd, MMRRC 036778) were crossed with 5XFAD transgenic mice (RRID: MMRRC_034840-JAX; overexpressing 5 gene mutations related to Familial Alzheimer’s Disease (FAD), 3 mutations in human APP (Swedish, K670N, M671L; Florida, I716V; and London, V717I) and 2 mutations in human presenilin 1, M146L and L286V). This cross resulted in a 5XFAD+/−/DBH-Cre+/− mouse line used for pathological endpoints. Female and male mice were used in these studies, and group sample sizes and the experimental designs are indicated in Fig. 1. To induce expression of inhibitory Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) on LC neurons, 5XFAD/DBH-Cre mice received bilateral LC injections of AAVs for expression of a Cre-dependent inhibitory DREADD (rAAV5-eF1a-DIO-hM4Di-mCherry) or mCherry as a control (rAAV5-eF1a-DIO-mCherry). The AAVs were obtained from the Stanford Gene and Virus Vector Core. Bilateral injections were administered at 2 loci targeting the LC (A/P -5.45 mm, M/L +/- 1.3 mm, D/V -3.8 mm & -3.4 mm) with 0.5 µL total per side. Following viral transfection, DBH promoter-dependent Cre recombinase expression resulted in transcription of the DREADD or the control mCherry fluorescent protein tag, specifically in NE neurons of the LC. To activate the DREADD, mice were dosed with the designer drug clozapine-N-oxide (CNO) at 2 mg/kg/day via subcutaneous pump administration for 1 month (for experimental timelines, see Fig. 1). Pumps (model 1004, Alzet, Cupertino, CA) loaded with CNO, were inserted subcutaneously in the back of each mouse under anesthesia.
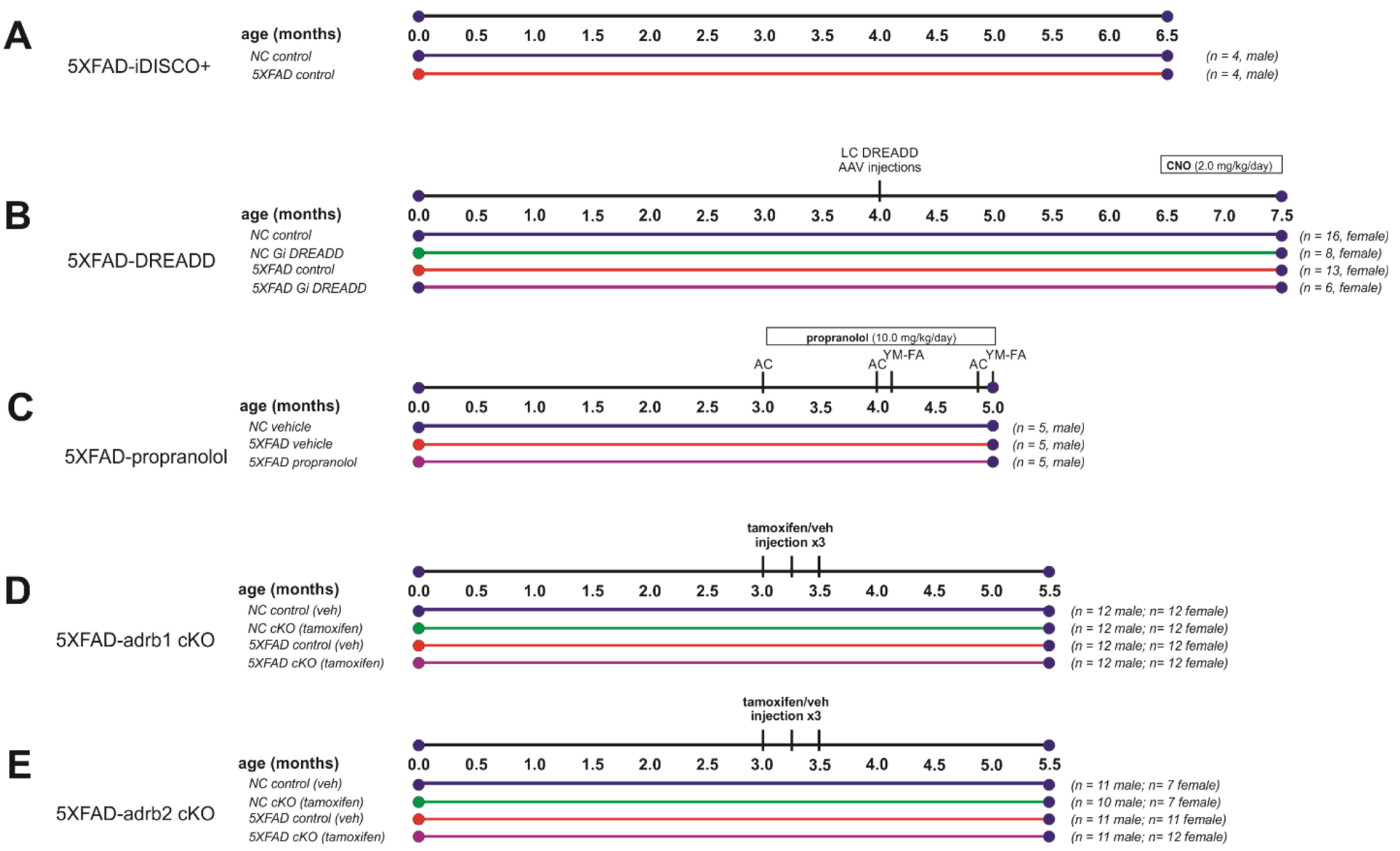
Fig. 1
Experimental Designs. (A) 5XFAD mice and non-carrier (NC) controls were aged to 6.5 months old for iDISCO + and light sheet imaging of brain pathology. (B-E) Inhibition of noradrenergic signaling was examined in parallel studies. (B) Immunological effects of the 5XFAD genotype and chemogenetic inhibition of locus coeruleus (LC) noradrenergic neurons with Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) were examined in 5XFAD and NC control mice. DREADD agonist clozapine N-oxide (CNO) was administered for 28 days via a pump to activate an inhibitory (Gi) DREADD expressed on LC noradrenergic neurons. Tissue was collected at 7.5 months of age. (C) Effects of beta-adrenergic receptor blockade with propranolol were examined in 5XFAD mice, and vehicle-treated 5XFAD mice and NC controls were also compared. Propranolol was administered daily for 2 months (10 mg/kg/day; intraperitoneal). Behavior was assessed in activity chamber (AC) and Y-Maze-forced alternation (YM-FA) assays. Tissue was collected at 5 months of age. (D-E) Effects of conditional deletion of adrb1 or adrb2 in microglia were studied in male and female 5XFAD and NC control mice. Three doses of tamoxifen or the vehicle (veh) control were administered to initiate the deletion of ADRB1 or ADRB2 on myeloid lineage cells, including microglia. Gene expression recovers in peripheral myeloid lineage cells with cell turnover but remains absent from microglia in the brain. Tissue was collected at 5.5 months of age
Propranolol studiesInitial body weight was used as a pseudo-randomization parameter for assigning mice to drug treatment groups (see Fig. 1 for Experimental Design). Three-month-old male 5XFAD mice (Jackson, see above) were administered the beta-adrenergic antagonist propranolol (10 mg/kg, i.p.; n = 5) or vehicle (i.p.; n = 5) daily for 2 months. A NC control group was dosed daily with the vehicle (i.p.; n = 5). An NC propranolol group was not included in this study as the aim was explicitly to determine the effect of propranolol on AD-related behavior and pathology in 5XFAD mice. Behavior was assessed in the Activity Chamber (AC) and Y-Maze: Forced Alternation (YM-FA).
Conditional knockout of ADRB receptors in microglia studiesFor conditional gene deletion studies, mice with a floxed Adrb1 sequence (gift of Steven Thomas, University of Pennsylvania; C57BL/6J background) or a floxed Adrb2 sequence (gift of Gerard Karsenty, Columbia; C57BL/6J background) were crossed with mice in which estrogen receptor ligand-binding-dependent expression of Cre recombinase (CreER) was linked to the expression of the C-X3-C motif chemokine receptor 1 (Cx3cr1) gene (Jackson 021160; B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/WganJ). These crosses resulted in transgenic mouse models (Adrb1-flox+/+/Cx3cr1-CreER+/− or Adrb2-flox+/+/Cx3cr1-CreER+/−) with tamoxifen-inducible genetic deletion of Adrb1 or Adrb2 specifically in myeloid lineage cells (e.g., microglia and macrophages). These lines were then crossed with 5XFAD hemizygous mice (Jackson, see above) to examine the effect of deletion of adrb1 or adrb2 specifically in myeloid lineage cells in 5XFAD mice from both sexes (for Experimental Design and N’s, see Fig. 1). To initiate gene deletion, mice were dosed with tamoxifen (Cayman Chemical 13258; 400 mg/kg; p.o.) or corn oil vehicle (Santa Cruz Biotechnology; sc-214761) using a 3-dose regimen, once per week, designed to maximize Cre-recombinase expression and genetic deletion across all myeloid lineage cells, including microglia. Persistent gene deletion specific to microglia was confirmed with RNAScope colocalization of allograft inflammatory factor 1 (aif1; ionized calcium-binding adapter molecule 1; iba1; microglia marker) with adrb1 or adrb2 in control mice but not in mice dosed with tamoxifen (See Supplemental Methods and Supplemental Figure S2).
Behavioral testingBehavior was tested in the propranolol study (Fig. 1). Mice were handled to obtain body weight and before the beginning of behavioral testing to habituate mice. On the days of behavioral testing, mice were moved to a holding area adjacent to testing rooms prior to the start of assessments. Dosing and tissue collection rooms were separate from housing and behavioral testing rooms. The dosing rooms and the holding area were lit by a red light. Food and water were freely available. All behavioral tests were performed and scored by an experimenter blind to experimental groups.
Activity chamber (AC)The AC assessed general locomotor activity and exploration as described previously [43, 44]. Mice were placed in one corner of a square Activity Arena (43 × 43 × 30 cm; Med Associates Inc., St. Albans, Vermont; Model ENV-515) located inside a dark sound-attenuated chamber (74 × 60 × 60 cm) and allowed to explore the arena freely. The movement was tracked by an automated tracking system with three planes of infrared detectors during a 10-minute trial. Parameters measured included distance moved, vertical counts (rearing), and vertical time in the periphery and center of the arena. The periphery was defined as the zone within 5 cm of the arena wall. The arena’s surface was cleaned with 1% Virkon disinfectant between each trial. AC was conducted pre- and post-dosing (CNO or propranolol) at 4- and 8-weeks post-dosing (3, 4, and 5 months of age; propranolol study). Daily dosing occurred at the end of the day after behavioral testing was completed.
Y-maze: forced alternation (YM-FA)The YM-FA was used to assess spatial memory. This test is based on the tendency of rodents to preferentially explore a novel environment over a familiar one. In this case, a rodent should prefer to explore a different arm of the maze than the arm they had previously explored. The maze was plastic with 3 arms in a “Y” shape (each arm 40 × 8 × 15 cm). The test consisted of two 8-minute trials separated by a 1-hour intertrial interval (ITI). To start each trial, mice were placed at the end of one of the arms (Start Arm). During the first trial (Training), mice were only allowed to explore two of the three arms (Familiar Arms). A plastic insert blocked off the third arm (Novel Arm). The Novel Arm was pseudorandomized to avoid any location bias. During the second trial (Testing), this insert was removed, and the mice were allowed to explore all three arms. The trials were recorded with an overhead camera and tracked with Ethovision XT (Noldus Information Technology, Wageningen, Netherlands). Between each trial, the surface of the maze was cleaned with 1% Virkon disinfectant. Y-maze was conducted at 4- and 8-weeks post-dosing (at 4 and 5 months of age).
Tissue collectionFor the propranolol study, mice were dosed with a final dose of propranolol 1 h prior to tissue collection. For terminal collection for the DREADD, propranolol, and conditional knockout studies, mice were deeply anesthetized with isoflurane. Prior to perfusion, whole blood was collected from the right ventricle via cardiac puncture (23 g needle) into lithium heparin-containing vials (BD microtainer plasma tubes, Becton Dickinson 365958) or K3EDTA-containing vials (propranolol study; Greiner Bio-One, MiniCollect Tube Reference #450475) for plasma collection. Plasma tubes were stored on ice before centrifugation within 60 min of collection. For perfusion, the right atrium was opened, and mice were transcardially perfused with ice-cold phosphate-buffered saline (PBS; 11.47 g sodium phosphate dibasic and 2.30 g sodium phosphate monobasic per 1 L deionized water; pH 7.4) through a 25 g needle. The perfused brain was removed. The brain was bisected coronally at the level of the mammillary bodies into the forebrain and hindbrain. The forebrain was hemisected. The left forebrain was immediately flash-frozen on dry ice and stored at -80 °C for later analysis. The right forebrain and hindbrain were post-fixed with 4% paraformaldehyde in a 15 ml conical centrifuge tube (48 h, 4 °C). Afterwards, the right hemispheres were rinsed 2 × 12 h in phosphate buffer (PB) and cryoprotected for at least 72 h (until sunk) in 30% sucrose in PB. Fixed brains were then rapidly frozen in isopentane on dry ice. All frozen tissue was stored at -80 °C. Whole blood was centrifuged (11 K rpm for 2 min) for plasma separation, and plasma was aliquoted and frozen on dry ice. Brain and plasma samples were stored at -80 °C.
Aβ ELISATo determine the amount of soluble and insoluble Aβ(1−40) (Aβ40) and Aβ(1−42) (Aβ42) present in brain tissue, cortical samples were processed as previously described [11, 33]. In brief, tissue dissections were weighed and homogenized in 10 volumes of tris-buffered saline (TBS) containing a protease inhibitor cocktail. The samples were centrifuged at 175,000 g for 30 min at 4 °C. The supernatant (TBS-soluble homogenate) was collected and kept at -20 °C. The pellets were re-homogenized in the same volume of TBS-T (TBS/1% Triton X-100 with protease inhibitor cocktail) at 4 °C, centrifuged at 175,000 g for 30 min at 4 °C, and the resultant supernatant (TBS T-soluble homogenate), containing membrane-bound Aβ, was collected and kept at -20 °C. Subsequently, the pellets were homogenized in ice-cold 5 M guanidine-HCl in 50 mM Tris (pH 8.0). The homogenates were mixed for 4 h at room temperature and used to measure insoluble Aβ40 and Aβ42. Finally, amounts of TBS-soluble and guanidine-soluble forms of Aβ were quantified using ELISA kits specific for human Aβ40 and Aβ42 (Invitrogen; #KHB3481 for Aβ40; #KHB3441 for Aβ42) following the manufacturer’s instruction. The final values were normalized to the amount of loaded wet tissue.
Multiplex mouse cytokine assayTissue cytokines were analyzed in brain homogenate from hippocampal (DREADD studies) or frontal cortex (conditional KO studies) dissections using a Luminex 48-plex mouse cytokine assay (ThermoFisher, EPX480-20834-901). The panel includes Pro-inflammatory cytokines: BAFF, G-CSF (CSF-3), GM-CSF, IFN alpha (IFNA), IFN gamma (IFNG), IL-1 alpha (IL1A), IL-1 beta (IL1B), IL-2, IL-6, IL-12p70, IL-17 A (CTLA-8), IL-18, IL-23, IL-27, IL-31, TNF alpha (TNFA); Anti-inflammatory cytokines: IL-4, IL-10, IL-13, IL-19; Cytokines involved in immune regulation and growth factors: IL-3, IL-5, IL-7, IL-9, IL-15/IL-15R, IL-22, IL-25 (IL-17E), IL-28, IL-33, LIF, M-CSF, RANKL; Chemokines: ENA-78 (CXCL5), Eotaxin (CCL11), GRO alpha (CXCL1), IP-10 (CXCL10), MCP-1 (CCL2), MCP-3 (CCL7), MIP-1 alpha (CCL3), MIP-1 beta (CCL4), MIP-2, RANTES (CCL5); Growth factors/regulators: Betacellulin (BTC), Leptin, VEGF-A; and Soluble receptors: IL-2R, IL-7R alpha (IL17RA), IL-33R (ST2). Briefly, tissue was homogenized in homogenization buffer containing proteinase inhibitor (1:100) by pulling it through a 23 g needle (15x) and then sonicating it with 3 × 3 s pulses. Homogenate was spun at 14,000 g for 10 min, and protein concentrations were determined using the Pierce BCA assay. Samples were diluted to a standard concentration of 6 µg/µL. Brain homogenates were run in singlets on a 96-well plate alongside a standard curve and quality control calibration samples.
ProteomicsIn the propranolol study, proteins from mouse hippocampal dissections were analyzed similarly to prior methods [34]. Proteomics analyses were performed at the Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University Mass Spectrometry (SUMS - RRID: SCR_017801). Lysis buffer (5% SDS, 50 mM TEAB, and 1X Protease and Phosphatase Inhibitors) was added to tissue samples, which were then homogenized using a bead mill. The lysate was cleared and transferred for filter-supported digestion. Proteins were reduced with 10 mM DTT at 550 °C for 30 min, followed by alkylation with 30 mM acrylamide for 30 min at room temperature. Trypsin/LysC protease (0.5 µg; Promega) was added to each sample for overnight digestion at 37 °C. After digestion, the reaction was quenched with 1% formic acid, and peptides were eluted and dried. Pierce Quantitative Fluorometric Peptide Assay kit (Thermo Fisher Scientific) was used for peptide quantification. The peptide mixture was dried using a speed vacuum centrifuge before dissolution in reconstitution buffer (2% acetonitrile with 0.1% formic acid). A total of 1 µg of peptides was used for subsequent LC-MS/MS analysis. Mass spectrometry was performed using an Orbitrap Eclipse Tribrid mass spectrometer RRID:022212 (Thermo Scientific, San Jose, CA, USA) with liquid chromatography using an Acquity M-Class UPLC (Waters Corporation, Milford, MA, USA). A flow rate of 300 nL/minute was used, where the mobile phase A was 0.2% formic acid in water, and the mobile phase B was 0.2% formic acid in acetonitrile. Analytical columns were prepared in-house with an inner diameter of 100 μm and pulled into to a nanospray emitter using a P2000 laser puller (Sutter Instrument, Novato, CA, USA). The column was packed using C18 Reprosil Pur stationary phase (1.8 μm particle size) to a length of ∼ 25 cm. Peptides were directly injected into the analytical column and separated using a gradient of 2–45% solvent B over 80 min, followed by a high-B wash. The mass spectrometer was operated in a data-dependent fashion using CID fragmentation for MS/MS spectra generation. For data analysis, the RAW data files were processed using Byonic v4.1.5 (Protein Metrics, Cupertino, CA, USA) to identify peptides and infer proteins. Proteolysis with Trypsin/LysC was assumed to be semi-specific, allowing for N-ragged cleavage with up to 2 missed cleavage sites. Precursor mass accuracies were held within 12 ppm and 0.4 Da for MS/MS fragments. Cysteine modified with propionamide was set as fixed modifications in the search. Proteins were held to a false discovery rate of 1%, using a standard reverse-decoy technique [45]. Pathway analysis for proteomics was conducted using the KEGG mouse 2019 dataset in Enrichr. All proteins showing 2-fold up- or down-regulation (t-test, p < 0.05) in response to the 5XFAD genotype or propranolol treatment were included in pathway analyses.
Immunohistochemistry for 6E10, iba1, and TH in brain slicesImmunohistochemistry was used to label and quantify ionized calcium-binding adapter molecule 1 (iba1), a marker for microglia/macrophages, Aβ (6E10), and tyrosine hydroxylase (TH), alongside a nucleic acid stain, 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI). Fixed brains were serially sectioned (at -18 °C using a Microm HM-550 cryostat) in a coronal plane, creating six series of sections spanning the rostrocaudal axis. For the forebrain, sections were 40 μm thick with 240 μm intervals between sections within each series, and for the hindbrain, sections were 30 μm thick with 180 μm intervals. Sections were stored in cryoprotectant buffer (30% ethylene glycol, 20% glycerol in 0.05 M phosphate buffer, pH 7.4). Multilabel fluorescent immunohistochemistry was performed on one series of brain sections to double-label iba1 and 6E10 in the rostral hippocampus (from 0.26 to -2.92 mm Bregma) [46] and on another series for TH labeling through the LC (from − 5.34 to -5.80 mm Bregma) [46]. Free-floating sections were incubated at room temperature in 24-well tissue culture plates gently shaken on an orbital shaker. All rinses were 15 min unless stated otherwise. Sections were rinsed three times in 0.05 M PBS, and then preincubated for 1 h in PBS containing 1% Triton X-100 (PBST) and 3% bovine serum albumin. Sections were incubated for 18 h with a goat anti-iba1 primary antibody (Abcam, ab5076, 1:1000), a mouse anti-6E10 primary antibody (Biolegend, 803001, 1:1000; binds to amino acid residues 1–16 of Aβ), or a chicken anti-TH primary antibody (Abcam, ab76442, 1:2000) in 0.3% PBST and 1% bovine serum albumin. Following 3 PBS rinses, sections were incubated for 2 h in IgG and/or IgY secondary antibodies, each diluted 1:250 in PBS (Cy5-conjugated AffiniPure donkey anti-chicken, 703-175-155; Cy3-conjugated AffiniPure donkey anti-goat, 705-165-147; 488-conjugated AffiniPure donkey anti-mouse, 715-545-151; Jackson Immunoresearch, Bar Harbor, ME). The secondary incubation included DAPI (D9542, Sigma-Aldrich, St. Louis, MO) diluted 1:5000. Free-floating sections were then rinsed 3 times in PB, mounted on clean glass slides, and allowed to air-dry to affix sections to slides immediately prior to coverslipping with polyvinyl alcohol mounting medium with DABCO antifade (10981, Sigma-Aldrich).
LC NE stereological cell countsLC NE cells were counted using stereology methods by an experimenter blind to treatment groups. Three sections, 180 μm apart, through the rostral to mid-rostrocaudal LC, were selected for counting. TH-positive neurons were counted via the optical fractionation method using a Zeiss AxioImager M1 microscope (Carl Zeiss) and Stereo Investigator software (version 2019.1.1, MBF Bioscience, Vermont). The sampling grid was set to cover 20% of the LC.
Whole-brain immunostaining, clearing, imaging, and quantificationImmunofluorescence staining and iDISCO+A separate cohort of male 5XFAD mice and NC control mice (Jackson Labs, RRID: MMRRC_034840-JAX) were used for brain-wide imaging studies. Mice were aged to 6.5 months before being anesthetized with 4% isoflurane and intracardially perfused with PBS followed by 4% PFA. Brains were removed, hemisected, and post-fixed overnight. Immunofluorescence staining and iDISCO+ were performed as previously described [47, 48]. The hemispheres were dehydrated in a methanol series, bleached in methanol with 5% hydrogen peroxide, rehydrated, permeabilized, and blocked. They were then incubated with primary antibodies for 7 days. In the left hemispheres, microglia and Aβ plaques were labeled with goat anti-iba1 (Abcam; ab5076; 1:100) and mouse anti-6E10 (Biolegend; 803001, 1:100) antibodies, respectively. In the right hemispheres, TH was labeled with rabbit anti-TH (Sigma Millipore; AB152; 1:200). After washing thoroughly, hemispheres were incubated with secondary antibodies for 7 days (Jackson Immuno Research: donkey anti-goat Cy3, 705-165-147, 1:100; donkey anti-mouse Cy5, 715-175-151, 1:100; donkey anti-rabbit Cy3, 711-165-152, 1:100). Tissue was repeatedly washed before being embedded in 1% low melting point agarose (ThermoFisher; R0801), dissolved in PBS with 0.02% sodium azide. The brains were dehydrated again, incubated in dichloromethane, and cleared/stored in dibenzyl ether.
Light sheet microscopy (LSFM)Cleared brains were imaged in 3D as described [48]. Briefly, brains were mounted with a c-clamp and immersed in ethyl cinnamate for imaging with a Zeiss Lightsheet 7. 3D images of entire hemispheres consisted of tiled z-stacks (∼ 800 × 688 pixels each), which were stitched together (10% overlap). Light sheets (10 μm thick) were pivot scanned and averaged together. A 2.5x detection objective captured images with 0.52x zoom (3.52 μm resolution). Z-steps were 5 μm for left hemispheres and 6 μm for right hemispheres. For concurrent imaging of autofluorescence and immunolabeling of 6E10 and iba1, autofluorescence was excited at 488 nm (10% power), and emissions passed through a 505–530 nm filter. Cy3 was excited with 561 nm light (30% power), and emissions passed through a 585 nm long pass filter. Cy5 was excited with a 638 nm laser (5% power), with emissions also passing through a 585 nm long pass filter. For TH staining, the 488 nm laser was set to 6% power, and emissions passed through a 505–545 nm filter. The 561 nm laser (2% power) evoked Cy3 fluorescence, passing through a 575–615 nm filter. The exposure time was 50 ms for all wavelengths.
Atlas registrationProcessing was automated using UNRAVEL [48]. The newer, Python-refactored code is available here (github.com/b-heifets/UNRAVEL/) with documentation here (b-heifets.github.io/UNRAVEL/index.html). In some instances, older scripts were used for this study, (github.com/b-heifets/UNRAVEL/blob/feature), with additional information here (github.com/b-heifets/UNRAVEL/blob/feature/Heifets_lab_guides/UNRAVEL_guide_Heifets_lab_021623.pdf). The autofluorescence channel of stitched z-stacks was downsampled 8x. Midline cuts were digitally corrected using 3D Slicer by trimming excess contralateral tissue and/or adding missing tissue to improve registration quality. Brain tissue was masked using the Ilastik project (pixel classification workflow) to zero out external voxels for registration at 50 μm resolution. Registration and warping were performed with modified scripts from MIRACL [49] using an iDISCO+/LSFM-specific average template brain [50] (25 μm resolution; Allen Mouse Brain Common Coordinate Framework v3 with region labels from 2017). Registration quality was visually inspected with ITK-SNAP.
Voxel-wise statisticsImmunofluorescence (IF) images were background-subtracted using the rolling ball method (pixel radius 20) to remove autofluorescence and normalize background intensities. TH-IF images were warped to atlas space using transformation matrices from registration. Due to the mouse origin of the 6E10 antibody, capillary staining varied with perfusion quality. Ilastik was trained to segment this non-specific staining in the raw IF images, and the resulting mask was applied to remove these artifacts in the full-resolution background-subtracted 6E10-IF images. Both non-reactive and reactive microglia were present in 5XFAD brains, with reactive microglia often aggregating around Aβ plaques. For voxel-wise analyses, Ilastik was trained to segment these aggregates and individual reactive microglia using raw iba1-IF training images. Ilastik was trained to identify reactive microglia using images from both control and 5XFAD brains (three 2D images per brain). Pixels of presumed reactive microglia and background (i.e., pixels not corresponding to reactive microglia) were sparsely labeled by the experimenter during training. Microglia were considered reactive either when they were aggregated (e.g., surrounding A-beta plaques) and/or iba1-immunoreactivity was higher than in the non-reactive microglia from the training set (e.g., in control brains). Ilastik training focused on minimizing the detection of resting microglia in control brains while maximizing the detection of reactive microglia in 5XFAD brains. Reactive microglia were segmented in raw iba1-IF images for all brains. Separately, autofluorescence was removed from each iba1-IF image by rolling ball background subtraction. To preserve signal from reactive microglia in the background-subtracted iba1-IF images, segmentations from Ilastik for each brain were used to zero out voxels not corresponding to reactive microglia. Masked and background-subtracted 6E10-IF and iba1-IF images were then warped to atlas space.
Atlas space images were individually z-scored using a brain mask that excluded the ventricles, olfactory bulb, and undefined regions. IF images were smoothed using fslmaths (100 μm kernel) from FSL (FMRIB software library). Voxel-wise comparisons were performed with FSL’s randomise_parallel command (18,000 permutations) using a t-test design. Adjustments to p-values for multiple comparisons were made with false discovery rate correction, defining clusters of significant voxels. The most stringent q value-producing clusters were used for each immunolabel (q < 0.2 for 6E10 and iba1 and q < 0.4 for TH). This corresponded to an adjusted p value threshold of 0.025 for 6E10 and iba1 and 0.011 for TH. Cluster extent thresholding filtered out small clusters likely representing noise, with empirically determined minimum cluster sizes of 400 voxels for 6E10 and iba1, and 100 voxels for TH. For TH clusters, masks of regions with dopaminergic or noradrenergic neurons were used for TH+ cell density measurements during cluster validation. Otherwise, the full extent of the clusters was used for label density measurements.
Cluster validationCluster validation was conducted as described [48]. To confirm that intensity-based differences in voxel-wise analyses reflect a difference in label or cell density, clusters were warped to tissue space and scaled to full resolution. Ilastik was trained to segment Aβ plaques in raw 6E10-IF images, reactive microglia in iba1-IF images, and TH+ fibers and cells in TH-IF images. Label densities in clusters were calculated for 6E10+ aggregates, reactive microglia, and TH+ fibers as ((segmented voxel volume) / (cluster volume)) * 100. TH+ cells were counted using the cc3d.connected_components() function with a connectivity of 6, and the cell count was divided by the cluster volume to calculate cell density. Clusters were considered valid if an unpaired, one-tailed t-test showed a significant difference in cell or label densities between groups. Images with valid clusters were visualized in 3D (DSI Studio). Regional composition was determined by multiplying binarized valid cluster images with the atlas and represented in sunburst plots (Flourish). Information on valid clusters is summarized in Supplemental Table S10, with region abbreviations defined in Supplemental Table S9. The top four regions’ volumes were calculated and reported if they constituted more than 80% of the total cluster volume; otherwise, subregions were collapsed into parent regions.
General statisticsUnless stated otherwise (see Methods - LSFM), statistical analyses were performed with GraphPad Prism 10.2. Repeated measures, one-way or two-way analyses of variance were followed by post-hoc comparison of select treatment groups with appropriate tests correcting for multiple comparisons. Significance was reported relative to p < 0.05, but results with effects approaching this threshold are also discussed as relevant trends [51, 52].
留言 (0)