
記住我



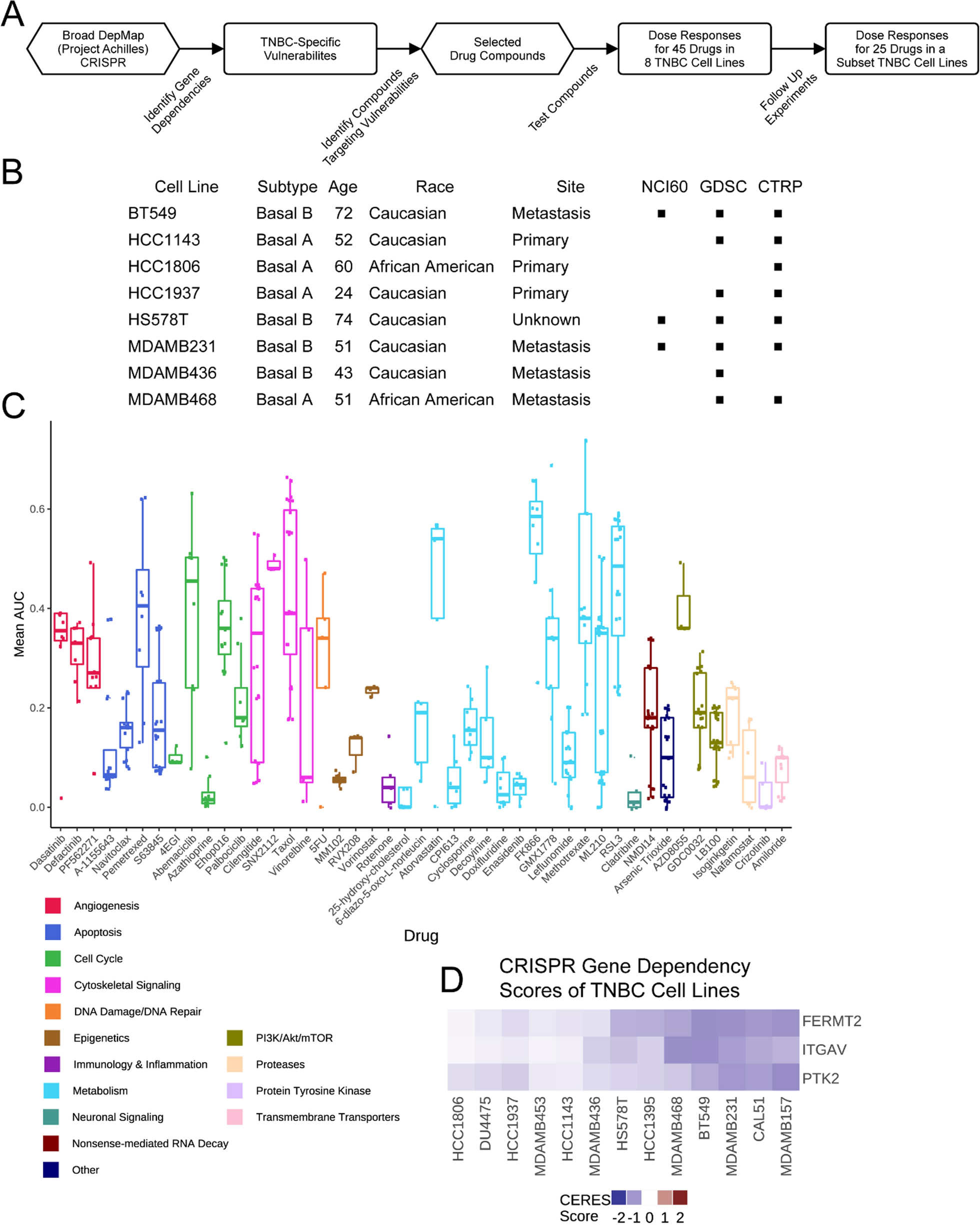
We queried the gene dependency data from the Broad DepMap dataset for genes with a variable range of dependency (i.e., vulnerabilities in at least two, but not all, breast cancer cell lines), enriched in TNBC cell lines compared to other breast tumor subtypes, with CERES dependency scores of -0.7 or lower. From these dependencies, we constructed a 45-drug panel composed of compounds that would inhibit either the gene product or related cellular process of those dependencies (Fig. 1A and Supp. Table 1 & 2). A diverse panel of eight cell lines representing basal A and basal B subtypes, derived from primary or metastatic sites, and including two African American-derived lines was assembled for our drug screening (Fig. 1B). Inclusion of these cell lines in other drug treatment datasets was noted (Fig. 1B, black square). Nine-point dose response curves were fitted and the area under the curve (AUC) was calculated (Supp. Table 3) for each cell line-drug pair such that higher AUC values correspond to higher drug sensitivity (i.e., 1-Viability area; see Methods). Notably, our dataset adds 18 drugs not previously tested in TNBC as part of large, public screening efforts (comparison via CellMinerCDB, which aggregates pharmacogenomics datasets from multiple institutes) complementing the existing body of work in this area [31].
Fig. 1
A drug treatment dataset designed using the DepMap applied to TNBC cell lines. A. Workflow of drug panel design and application. B. Description of cell lines used to generate the drug treatment dataset, including their subtype, patient’s age and race, the site of collection, and the presence (black square) of the cell line in other drug screening datasets. C. AUC values of all screened drugs are shown as a boxplot which displays a five-number summary, including the minimum, first quartile, median, third quartile, and maximum AUC, color-coded by mechanism of action. D. Cell line gene dependency values from the DepMap for FERMT2, PTK2, and ITGAV are shown. Dependency scores of -0.7 or lower are considered dependent
The tested compounds produced a range of responses across the cell lines (Fig. 1C, see Methods). Several drugs had a consistent effect across all cell lines (e.g., dasatinib, A1155643, MM102, vorinostat, etc.), meaning the AUC values were tightly grouped, not exhibiting a differential response that would be amenable for biomarker identification. The unanticipated lack of variable responses to these compounds may relate to the time scale and specificity of inhibition using CRISPR instead of a drug. Other drugs, however, showed a wide range of AUC values (e.g., pemetrexed, abemaciclib, cilengitide, vinorelbine, methotrexate, and ML210) demonstrating that gene dependencies can help identify targetable pathways and processes that represent cell vulnerabilities. This group of compounds is more amenable to statistical analysis to identify biomarkers that might stratify patients by benefit. We performed validation studies on the 25 drugs that showed the greatest heterogeneity in our primary screen and found that cilengitide, an inhibitor of the ITGAV:ITGB3 integrin heterodimer, had a robust, reproducible phenotype across experiments (Spearman correlation between replicates = 0.67).
Cilengitide was included in our drug screen to target observed dependencies (based on the CERES metric developed by the Broad Institute [8]) on three proteins involved in cell adhesion: FERMT2, ITGAV, and PTK2. Seven out of 13 TNBC cell lines were dependent on FERMT2, which is also known as kindlin-2, an integrin co-activator that mediates signaling between integrins and the focal-adhesion kinase pathway (upstream of PI3K/AKT/mTOR) [32,33,34,35]. Of the FERMT2-dependent lines, six were also dependent on ITGAV and four of those were dependent on PTK2, the gene that encodes the focal adhesion kinase (Fig. 1D), suggesting that some TNBC cell lines are especially dependent on the focal adhesion complex involved in cell adhesion. Given our observations of the differential dependencies in DepMap and drug responses in our dataset, we chose to use a systematic approach to understand TNBC cell response to ITGAV inhibition by cilengitide.
TNBC cell lines exhibit differential sensitivity to the integrin inhibitor cilengitideWe grouped cell lines that had an IC50 value below 5 μM as sensitive (BT549, HS578T, MDAMB436, MDAMB468), while cells with an IC50 value over 5 μM were classified as resistant (HCC1806, HCC1937, HCC1143, MDAMB231), resulting in two equal groups of cell lines (Fig. 2A). Using conventional methods of measuring cell line drug sensitivity, area-under-the-curve (AUC) values were computed (Fig. 2B). Since growth rate is known to confound comparative drug sensitivity studies across cell lines [36], Growth Rate inhibition values (GR) were calculated and compared to the AUC. We found that both the area-over-the-curve (AOC; Fig. 2C) and AUC demonstrated striking resistance in HCC1937 and HCC1806 lines and placed BT549 and HS578T as among the most sensitive lines. An examination of cell morphology across the cell lines after 24 h of treatment revealed that BT549, HS578T, and MDAMB436 detached in the presence of cilengitide, whereas HCC1143, HCC1806, and HCC1937 all remained attached (Fig. 2D). Thus, while the IC50, AUC, and AOC metrics for line HCC1143 presented a mixed phenotype, the line’s continued attachment (Fig. 2D) and proliferation (Supp. Figure 1A) in the presence of cilengitide, in contrast to other sensitive lines (Supp. Figure 1B), led to its classification as a resistant cell line. To assess how consistent the sensitive and resistant cell line classifications would be with ITGAV inhibition, we examined TNBC cell proliferation in the presence of the small molecule, pan-ITGAV inhibitor GLPG0187, which was also well-tolerated in phase I trials [21]. We found that the cells’ designation of sensitive and resistant to cilengitide was consistent with response to GLPG0187 (Supp. Figure 1C and D).
Fig. 2
Some TNBC cell lines are dependent on adhesion proteins and respond to the integrin inhibitor, cilengitide. A & B. Dose response curves were fitted based on 9 doses of cilengitide for 8 TNBC cell lines. Representative results from 1 of 4 experiments are shown (A). The AUC values from replicate experiments together with the mean (horizontal bar) were plotted in order from most to least sensitive (B). C. GR metrics were calculated for the 8 TNBC cell lines treated with cilengitide, and AOC values from replicate experiments were plotted together with the mean (horizontal bar). Higher AOC values indicate increased sensitivity. D. Bright field images showing the morphology of cells at 24 h post-treatment with DMSO or cilengitide (5 µM) are shown (scale bar = 200 μm)
To understand the nature of the cell death induced by cilengitide in the sensitive lines, we cultured them in the presence of a caspase 3/7-activatable fluorescent dye. If cells engaged the caspase-mediated apoptotic pathway following detachment, it would indicate that they are undergoing anoikis [37]. Indeed, the sensitive cell lines all underwent detachment and showed caspase 3 activation by twelve hours or earlier, suggesting that the sensitive cells undergo apoptotic cell death in response to cilengitide-induced suspension (Supp. Figure 1E). Indeed, we noticed that in lines BT549 and HS578T, the detachment and fluorescence occurred even more rapidly, making it difficult to establish the order of events.
The dependencies on PTK2, FERMT2, and ITGAV observed in the DepMap had suggested that certain TNBC cell lines would be sensitive to inhibition of integrin-mediated adhesion. To assess whether the CRISPR vulnerabilities followed the same pattern of cilengitide sensitivity observed across the cell lines, we checked the sensitive and resistant cell line CRISPR dependencies from the DepMap. Of the three genes, ITGAV had the most striking difference in the CERES scores between the resistant and sensitive lines tested, though it did not reach significance (Supp Fig. 1F). Just as PTK2 dependency did not follow cilengitide sensitivity, FAK inhibition also failed to correlate with this characteristic. Two different FAK inhibitors (PF-562271 and GSK2256098C) in the Genomics of Drug Sensitivity in Cancer (GDSC) cell line drug screening database showed no relationship with cilengitide sensitivity (-0.39 and -0.18, respectively and neither was statistically significant using the available samples, Supp. Table 4). Unlike PTK2 or FERMT2, ITGAV is a direct target of cilengitide [12, 13]. Thus, the observed dependency on ITGAV largely translated to sensitivity to ITGAV inhibition in TNBC separate from the downstream effectors PTK2 and FERMT2.
Proteins involved in cell adhesion are differentially abundant between cilengitide sensitive and resistant linesCurrently, there exist contradictory reports regarding the role of target integrin expression and the response to cilengitide. In a retrospective study using patient tissues from the CORE and CENTRIC trials, which tested the efficacy of cilengitide in glioblastoma, the expression of target integrins was associated with clinical response to cilengitide: improved progression-free survival was reported in glioblastoma patients whose tumors expressed ITGAV:ITGB3 [38]. However, a separate in vitro study showed that breast cancer cell lines with higher expression of ITGB3 were resistant and could be sensitized by knockdown of ITGB3 to lower levels [16]. These data raised the possibility that a cilengitide-sensitive cell will express the target integrins at levels allowing effective inhibition.
Thus, we examined cilengitide target expression in two proteomics datasets: the BR80 [39] and CCLE [40], which together covered all of our cell lines and more integrins than either dataset alone. We calculated z-scores for the abundances provided in each dataset to facilitate comparison. The z-scores of the target integrins were added together to calculate an overall summary score and ranked from highest to lowest (Fig. 3A). In the BR80 dataset, the resistant line HCC1143 had the highest abundance of ITGAV and ITGB5, followed by the sensitive line HS578T. Furthermore, the cell lines with intermediate expression were all from the resistant group. In the CCLE dataset, which lacked the sensitive line HS578T, the resistant lines HCC1143 and MDAMB231 ranked highest for the z-score sum of ITGAV, ITGB3, and ITGB5 (Fig. 3B). Meanwhile, HCC1806 and HCC1937, which had the highest IC50 and GRMax response values, had lower expression (i.e., lower overall summary score) of the cilengitide targets than the sensitive line BT549, but higher expression than two other sensitive lines. To further validate these findings and to assay ITGB3 expression (no data in BR80) in HS578T cells (no data in CCLE), we performed flow cytometry and immunoblotting experiments to check integrin abundance. Using an antibody against the extracellular portion of the ITGAV:ITGB3 dimer, we could show that there was no significant difference in surface expression of this dimer between sensitive and resistant lines, and that HCC1143 and HS578T had the highest levels of this dimer (Fig. 3C). Immunofluorescent ITGAV:ITGB3 dimer staining confirmed similar punctate staining across cell lines (Supp. Fig. 2). Blotting experiments further supported the observation that HCC1143 was the highest ITGAV-expressing line (Fig. 3D and E), in agreement with the BR80 and CCLE datasets. Meanwhile, the resistant line HCC1143 and the sensitive line HS578T were the top expressors of ITGB3. Taken together, we found no relationship between cilengitide response and ITGAV or ITGB3 abundance, suggesting that at the protein level neither ITGAV nor ITGB3 could predict TNBC response to ITGAV inhibition.
Fig. 3
Elevated cell adhesion proteins are a distinguishing characteristic of cilengitide resistant and sensitive lines. A & B. Bar graphs of summed z-scores ranked from the highest to lowest sum for the cilengitide target integrins ITGAV and ITGB5 from the BR80 (A) and ITGAV, ITGB3, and ITGB5 from the CCLE (B) proteomics datasets are shown. C. Surface-expressed ITGAV:ITGB3 was assessed using flow cytometry and percentages of positive cells were quantified for each cell line, then plotted by sensitivity to cilengitide (left) or by cell line (right). No significant (ns) difference was found (t-test) between the sensitive and resistant lines. The mean with S.E.M. is shown. D. Protein levels of cilengitide targets ITGAV and ITGB3 were probed by immunoblot, with beta-actin (same blot as in Fig. 4F) serving as a loading control. Cell lines were loaded from left to right by their cilengitide sensitivity (most to least resistant). E. Three independent cell protein lysates from each cell line were quantified for ITGAV and ITGB3 and normalized to beta-actin. Resistant lines are colored in red, while sensitive lines are blue. Mean with S.E.M. is shown. F. TNBC-associated proteins (DisGeNET) were assessed for differential protein abundance (p < 0.05, unadjusted Wilcoxon signed rank) in the BR80 dataset between resistant (red) and sensitive (blue) lines and the abundance of the 7 proteins meeting the cutoff is shown as a heatmap with higher abundance in red and lower in blue. G. Differential protein abundance of the resistant and sensitive cell lines was determined across the entire BR80 proteomics dataset and used to perform gene set enrichment analysis (see Methods). The -log of the p-value is plotted for pathways that reached significance (p < 0.01)
Next, we evaluated more broadly protein expression patterns that may account for cilengitide resistance. We identified 22 proteins associated with TNBC (taken from DisGeNET, one of the largest databases of genes associated with human disease) that were present in the BR80 dataset and differentially abundant (unadjusted p < 0.05) between the sensitive and resistant lines (Fig. 3F). Among the identified proteins were ITGA6 and ITGB4; while neither is reported to be a target of cilengitide, it raised the possibility that integrins other than ITGAV and ITGB3 may modulate cilengitide sensitivity. Another of the TNBC-specific proteins identified, SRC, lent support to the hypothesis that upregulation of adhesion complex members may decrease cell response to cilengitide. To explore this possibility further, we performed GSEA using the MSigDB Hallmark signatures together with KEGG and WikiPathway adhesion gene set signatures on all differentially abundant proteins in the BR80 dataset across the two groups of cell lines. This analysis showed that proteins in the WikiPathways Focal Adhesion and Integrin-mediated Adhesion as well as the KEGG Focal Adhesion were enriched in the resistant cell lines (Fig. 3G). Furthermore, the leading-edge genes (i.e., genes contributing the most to the enrichment) in our analysis (Supp. Table 5) consisted of ECM proteins (e.g., FBN1, LAMB3, and COL4A2), integrins (e.g., ITGA2, ITGA3, ITGA6, ITGB1, and ITGB4), and proteins involved in the focal adhesion complex and its downstream signaling (e.g., ZYX, SRC, RAC1, ROCK1). These results indicated that differences in cell adhesion between sensitive and resistant cells mediate the response to ITGAV inhibition.
Integrin and ECM protein abundance in TNBC cell linesGiven the implication of integrin-mediated cell adhesion as the differentiator in ITGAV inhibition response, we probed the BR80 proteomics dataset, specifically examining the integrins and ECM proteins. The sensitive line HS578T showed marked upregulation of many ECM proteins (Fig. 4A), especially FN1, which was confirmed using immunocytochemistry (Fig. 4B). However, no other sensitive line exhibited this phenotype, making it unlikely to explain cell response to cilengitide. Overall, the resistant lines had a tendency toward lower abundance of ECM proteins compared to sensitive lines (Fig. 4A). A more consistent sensitive-resistant line difference was observed for integrin expression. The resistant lines expressed more integrins and at higher levels than the sensitive lines (Fig. 4C–E), with many of the integrins trending towards a significantly higher expression in resistant lines compared to sensitive lines. In addition to the earlier-noted ITGA6 and ITGB4, which were significantly upregulated in resistant lines, ITGA2, ITGA3, and ITGB1 trended towards significantly higher expression in resistant lines (Fig. 4C). The CCLE dataset also showed increased integrin abundance in resistant lines (Supp. Figure 3A). Flow cytometry was used to validate significantly higher cell surface ITGB4 expression in resistant, though not sensitive lines (Fig. 4F). Additionally, we performed immunoblotting of cell lysates for several integrins as well as the focal adhesion kinase (FAK) which is involved in mediating cell attachment through integrins [41,42,43]. In our blotting experiments, we found that FAK was consistently abundant in all cell lines (Fig. 4G, Supp. Figure 3B). There was an apparent trend towards ITGA6 upregulation in resistant lines though the increase in ITGA3 was less clear in our immunoblots than it was in proteomics (Fig. 4G, Supp. Figure 3B); in contrast, ITGB4 was clearly higher in resistant lines (Fig. 4G–H), consistent with the proteomics data. We additionally visualized ITGA6 and ITGB4 across the cell lines to confirm anticipated localization of these integrins to sites of cell adhesion (Supp. Figure 3C).
Fig. 4
Proteomic characterization of TNBC cell lines used in drug screen. A. A heatmap shows proteomics data of ECM protein expression in resistant (red) and sensitive (blue) TNBC cell lines. B. Immunofluorescence of FN1 (green) counterstained with DAPI (blue) in resistant (HCC1806, HCC1937) and sensitive (HS578T, BT549) cell lines. Scale bar = 200 µm. C. Integrin protein expression in resistant (red) and sensitive (blue) breast cancer cell lines from the BR80 proteomics dataset. The integrins targeted by cilengitide are labeled with yellow boxes. The p-value (unadjusted Wilcoxon signed rank) of differential abundance between resistant and sensitive lines is shown, with more significant p-values in darker purple. D. The z-score values from (C) were summed across rows for the available integrins and plotted to show relative integrin abundance across the cell lines. E. The z-scores from integrins in the CCLE were summed across cell lines and plotted as in D. F. Surface-expressed ITGB4 was assessed using flow cytometry and percentages of positive cells were quantified for each cell line, then plotted by sensitivity to cilengitide (left) or by cell line (right). A significant difference was found (t-test) between the sensitive and resistant lines (p < 0.001). The mean with S.E.M. is shown. G. Lysates were made from untreated cells (arranged from most to least sensitive; see triangle), and subjected to immunoblotting for several integrins, with beta-actin serving as a loading control. ITGA6 and ITGB4 were probed on the same membrane as ITGAV and ITGB3 in Fig. 3D (the same actin band appears in both figures). ITGA3 and FAK were run on a parallel blot and have a separate actin loading control. H. Three independent cell protein lysates from each cell line were quantified for ITGB4 and normalized to beta-actin. Resistant lines are colored in red, while sensitive lines are blue. The mean with S.E.M. is shown
We summarized our proteomics findings in graphs (Supp. Figure 4A) showing sensitive and resistant cell line integrin abundance in the context of their integrin binding partners and ECM proteins. This approach highlighted a few key differences in these two groups of cells. First, the ITGAV and ITGB5 monomers were more abundant in the sensitive lines compared to resistant lines, but only resistant lines showed a positive correlation between these proteins, suggesting a coordinated expression of these proteins that would result in increased dimer presence. The increased presence of this secondary cilengitide target on resistant cells could either represent a vulnerability because it would enable targeting by cilengitide, or a resistance mechanism because of the increased ratio of integrin target to cilengitide molecule. Given the cell phenotype observed, the latter possibility may be more likely. Second, ITGA6 and ITGB1 are positively correlated in lines sensitive to ITGAV inhibition mediated by cilengitide and GLPG0187, but not in the resistant lines, suggesting this pair may be a key laminin-binding mechanism in sensitive cells while resistant lines may rely more heavily on the ITGA6:ITGB4 integrin pair for laminin binding. However, ITGB1 abundance is lower in the sensitive lines compared to resistant lines, suggesting that it would be less effective at mediating adhesion to laminin than ITGA6:ITGB4 in the resistant lines. Thus, the sensitive lines would not have an alternative integrin-mediated attachment in the presence of ITGAV inhibition while resistant cells would, based on the presence of integrin monomer abundance and their correlations in the cell lines.
To explore the possibility of ITGA6:ITGB4 dimers representing a compensatory mechanism in the face of ITGAV blockade, we examined the correlation of protein abundance from the BR80 in complexes from the CORUM database that include these two integrins together with their ECM binding partner, laminin [44]. We reasoned that cells utilizing these specific complexes would express the components in a coordinated fashion. We found that pairs of ITGA6:ITGB4 complex members were more highly positively correlated using protein abundance in the BR80 across the ITGA6:ITGB4:Laminin 10/12 complex in resistant cell lines than in sensitive lines (Supp. Figure 4B), suggesting this complex is present and active in the resistant, but not sensitive, lines. Thus, different integrin and ECM repertoires might form the basis of resistant line continued attachment in the presence of cilengitide or GLPG0187.
Given the high expression of ITGA6, ITGA3, and ITGB4 in resistant cells relative to sensitive cells, we performed knockdown experiments with siRNAs targeting each of these integrins to determine their role in mediating cilengitide resistance. We did not observe increased sensitivity in resistant cells with knockdown of any of the integrins or in the presence of a blocking antibody to ITGB4 (Supp. Figure 5). While select knockdown of integrins failed to sensitize resistant cells to cilengitide, we cannot rule out the possibility that integrin crosstalk (e.g., a shift of an integrin monomer pairing with an alternative partner when another is knocked down [17, 45, 46] or compensatory upregulation [47, 48]) was the cause of persistent cilengitide resistance in the context of ITGA6, ITGA3, and ITGB4 knockdown.
Both laminin and fibronectin can confer cilengitide resistance to sensitive cellsThe targets of cilengitide, integrin dimers ITGAV:ITGB3 and ITGAV:ITGB5, bind RGD motifs found in ECM components such as fibronectin, vitronectin, and fibrinogen [12, 13]. Collagen and laminin are bound by other groups of integrins, including many integrin dimers formed by the ITGB1 subunit and the ITGA6:ITGB4 dimer [49], which are not targets of cilengitide. Thus, cilengitide sensitivity of a cell might be impacted by the available ECM substrates. To explore this possibility, we plated resistant cell lines and allowed them to deposit matrix for 2–3 days under normal growth conditions before decellularizing the plates and seeding sensitive lines on the deposited ECM proteins. The sensitive lines were allowed to adhere overnight then treated with cilengitide. The resistant-cell ECM proteins rendered sensitive lines resistant to cilengitide (Fig. 5A, top row; Supp. Figure 6A). To test whether increased matrix protein deposition may be the cause of the newly acquired resistance, each sensitive line was plated on ECM proteins deposited from each sensitive line (i.e., BT549 was plated on its own ECM as well as HS578T ECM, and MDAMB436 ECM). We found that sensitive-line ECM proteins did not confer cilengitide resistance, with the exception of the matrix proteins from line HS578T for MDAMB436 cells (Fig. 5A, bottom row; Supp. Figure 6A). Also, the ECM of sensitive lines did not confer sensitivity to resistant lines (Fig. 5B; Supp. Figure 6A). These results suggest that the ECM deposited by cancer cells can dictate response to cilengitide.
Fig. 5
Cilengitide sensitivity can be modulated by ECM. A. Sensitive lines were plated in triplicate on ECM deposited by resistant cells (top row; HCC1143 ECM in black, HCC1806 ECM in red, HCC1937 ECM in blue) or sensitive cells (bottom row; BT549 ECM in black, HS578T ECM in red, MDAMB436 ECM in blue). Cells were then treated with DMSO (closed circle) or 5 μM cilengitide (open circle) and their confluence was monitored at 4 h intervals for 60 h. For comparison, cells were also plated on untreated plastic (green curves). B. Resistant lines were plated on ECM deposited by sensitive cells (bottom row; BT549 ECM in black, HS578T ECM in red, MDAMB436 ECM in blue), treated with DMSO or 5 μM cilengitide, and their confluence was monitored at 4 h intervals for 72 h. For comparison, cells were also plated on untreated plastic (green curves). C. Sensitive lines BT549 and HS578T and resistant lines HCC1806 and HCC1937 were plated in triplicate on fibronectin (black), laminin (red), collagen I (green), or plastic (blue), treated with DMSO or 5 μM cilengitide, and their confluence was measured at 4 h intervals for 72 h. In all cases, a representative experiment is shown of 3 (A & B) or 2 (C) biological replicates
While plating cells on different ECM deposits, we noted that the ECM deposited by the resistant cell lines induced altered cell morphology in some sensitive lines (Supp. Figure 6B), though not the other way around (Supp. Figure 6C). Given that the resistant ECM converted sensitive cells to resistant cells, we aimed to characterize these morphological changes to see if they predicted cilengitide sensitivity. For example, cilengitide-sensitive MDAMB436 cells acquired a flattened morphology on HCC1806 and HCC1937 matrices, compared to its morphology on all three sensitive cell matrices, where it formed round cells with multiple projections (Supp. Figure 6B). Indeed, the matrix from cilengitide-resistant HCC1806 caused spreading in all three sensitive lines. However, while cilengitide-resistant HCC1937 ECM conferred resistance to all sensitive lines, it only induced morphological changes for BT549 and MDAMB436. Similarly, cilengitide-resistant HCC1143 ECM rendered all three sensitive lines resistant, but only HS578T acquired a different appearance. Thus, cell morphology changes were not always predictive of cilengitide response.
To better understand which ECM proteins may confer resistance to cells, we plated cells on laminin, collagen, or fibronectin. As was seen on cell line ECM, there were morphological shifts of sensitive cells on the different ECM substrates. Resistant lines also exhibited some morphology changes, though cilengitide-resistant HCC1806 remained largely unchanged regardless of ECM protein (Supp. Figure 6D). All tested lines were resistant to cilengitide treatment when plated on laminin or fibronectin (Fig. 5C). Collagen I also conferred resistance, except to cell line BT549 (Fig. 5C). Taken together, these results show that multiple ECM proteins can modulate cilengitide sensitivity but are only a part of what makes a cell sensitive to cilengitide.
Luminal breast cancer cells with lower integrin expression are sensitive to cilengitideConsidering the results from our integrin knockdown experiments showing that integrin-abundant lines are not sensitized to cilengitide, and the observation that cilengitide resistant cells express higher levels of multiple integrins, we sought to test whether cell lines expressing lower overall integrin levels might be sensitive to cilengitide. We reasoned that such integrin low cells would lack compensatory binding from integrins not targeted by cilengitide. Thus, when the dimers targeted by cilengitide (e.g., ITGAV:ITGB3) would be inhibited, this type of cell would be dislodged from the matrix. Previous studies in the normal mammary epithelium have shown that basal breast epithelial cells express more integrins, including the ITGA6:ITGB4 dimer, than luminal epithelial cells [45, 50]. Hence, we checked the integrin repertoire expressed by several luminal lines in the BR80 dataset. In line with the biology of the normal mammary epithelium, we found that luminal breast cancer cell lines expressed lower abundance of integrins than TNBC lines, with an especially striking difference between luminal and resistant TNBC lines (Fig. 6A, Supp. Figure 7A–B). Also, in the BR80, we noted additional integrin-low TNBC lines (e.g., CAL51, CAL148) and integrin-high TNBC lines (e.g., HDQP1, HCC38). The overall elevated integrin expression in basal tumors over luminal tumors was confirmed in patient data from the TCGA (Fig. 6B). Basal tumors also tended to express more ITGA6 and ITGB4 than luminal tumors, similar to our observations made in the cell lines (Fig. 6C and D). By employing the same approaches used with TNBC cell lines, we confirmed that the six tested luminal lines (EFM19, HCC1428, MCF7, MDAMB175VII, T47D, and ZR-75–1) exhibited the hallmarks of cilengitide sensitive lines, such as low AUC values, absent or suppressed growth, and cell detachment in the presence of cilengitide (Fig. 6E–G, Supp. Figure 7C). Interestingly, fibronectin rescued proliferation in four of the six lines and laminin rescued in only two of the six lines, in contrast to the universal rescue seen with both matrix proteins in TNBC lines (Supp. Figure 7D), possibly because different integrin repertoires will result in differential pathway activation even on the same substrate [51]. These data support the hypothesis that decreased overall integrin expression indicates increased cilengitide sensitivity and suggest that, in addition to utility in a subset of TNBC patients, cilengitide, or other ITGAV inhibitors, may be effective in luminal breast cancers as a whole.
Fig. 6
Breast cancer cell lines expressing lower levels of integrins are more sensitive to cilengitide. A. A heatmap of integrin protein expression in luminal (orange) and TNBC breast cancer cell lines from the BR80 proteomics dataset is shown. Known resistant lines are marked in red, sensitive lines in blue and lines with untested cilengitide response were marked in green. Z-score values were summed across rows to calculate overall abundance for the available integrins (magenta higher and cyan lower abundance). B–D. Density plots show the sum of overall integrin expression scores (B), or ITGA6 (C) or ITGB4 (D) protein abundance for breast cancer patient data, plotted by subtype (Basal = blue, Luminal A = yellow, Luminal B = red), from the METABRIC dataset. E. Luminal cell line dose response curves were fitted based on a 9-point titration of cilengitide. One of 3 representative experiments is shown. HCC1806 and BT549 are included for comparison. F. The AUC values for luminal lines (orange symbols) were compared with AUC values of a resistant (HCC1806; red) and a sensitive (BT549; blue) cell line. G. The AOC values for luminal lines (orange symbols) were compared with AOC values of a resistant (HCC1806; red) and a sensitive (BT549; blue) cell line. H. CAL51 (black) and known-sensitive line BT549 (blue) were plated in triplicate, treated with DMSO (circle, solid line) or 5 μM cilengitide (open circle, dotted line) and their confluence was monitored at 4 h intervals for 72 h. One of three representative experiments is presented. CAL51 cell morphology 72 h after treatment with cilengitide or DMSO is shown. I. HDQP1 (black) and known-sensitive line BT549 (blue) were plated in triplicate, treated with DMSO (circle, solid line) or 5 μM cilengitide (open circle, dotted line) and their confluence was monitored at 4 h intervals for 72 h. One of three representative experiments is presented. HDQP1 cell morphology 72 h after treatment with cilengitide or DMSO is shown. Scale bars = 100 μm
Our collective findings suggested that overall integrin expression may predict cilengitide sensitivity better than a single integrin. To further test the potential of using integrin abundance, we sought to perform drug testing in a set of TNBC cells that were not included in our original set of lines. Of the TNBC cell lines not yet tested, we found that the integrin-low line CAL51 (Fig. 6A) showed reduced growth and cell rounding in the presence of cilengitide similar to the sensitive line BT549 (Fig. 6H). Meanwhile, integrin-high HDQP1 (Fig. 6A) cells proliferated in the presence of drug and their morphology was unchanged (Fig. 6I). Thus, our hypothesis that integrin-abundant cell lines would be resistant to cilengitide (Fig.
留言 (0)