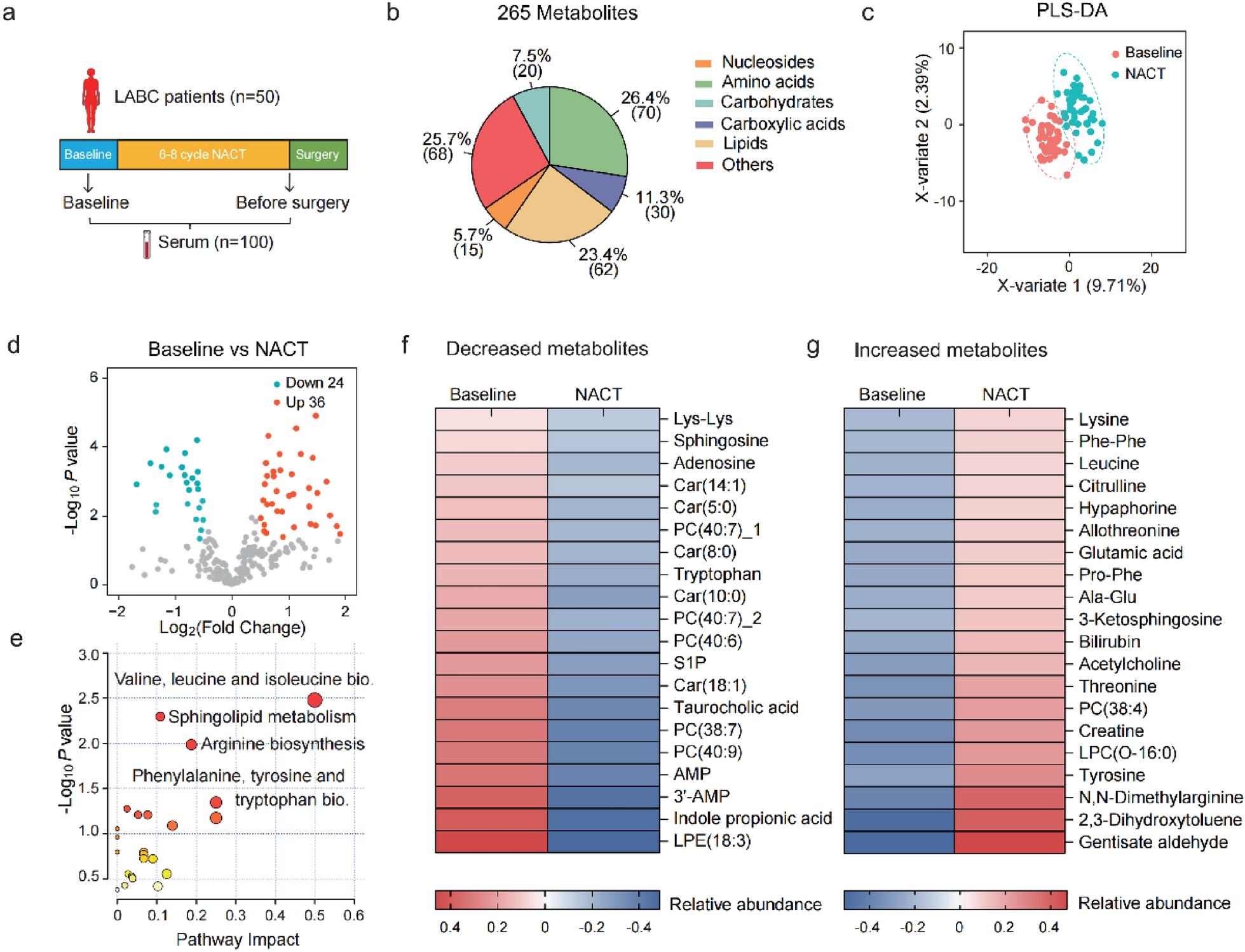
MiRome analysis on patients’ serum samples
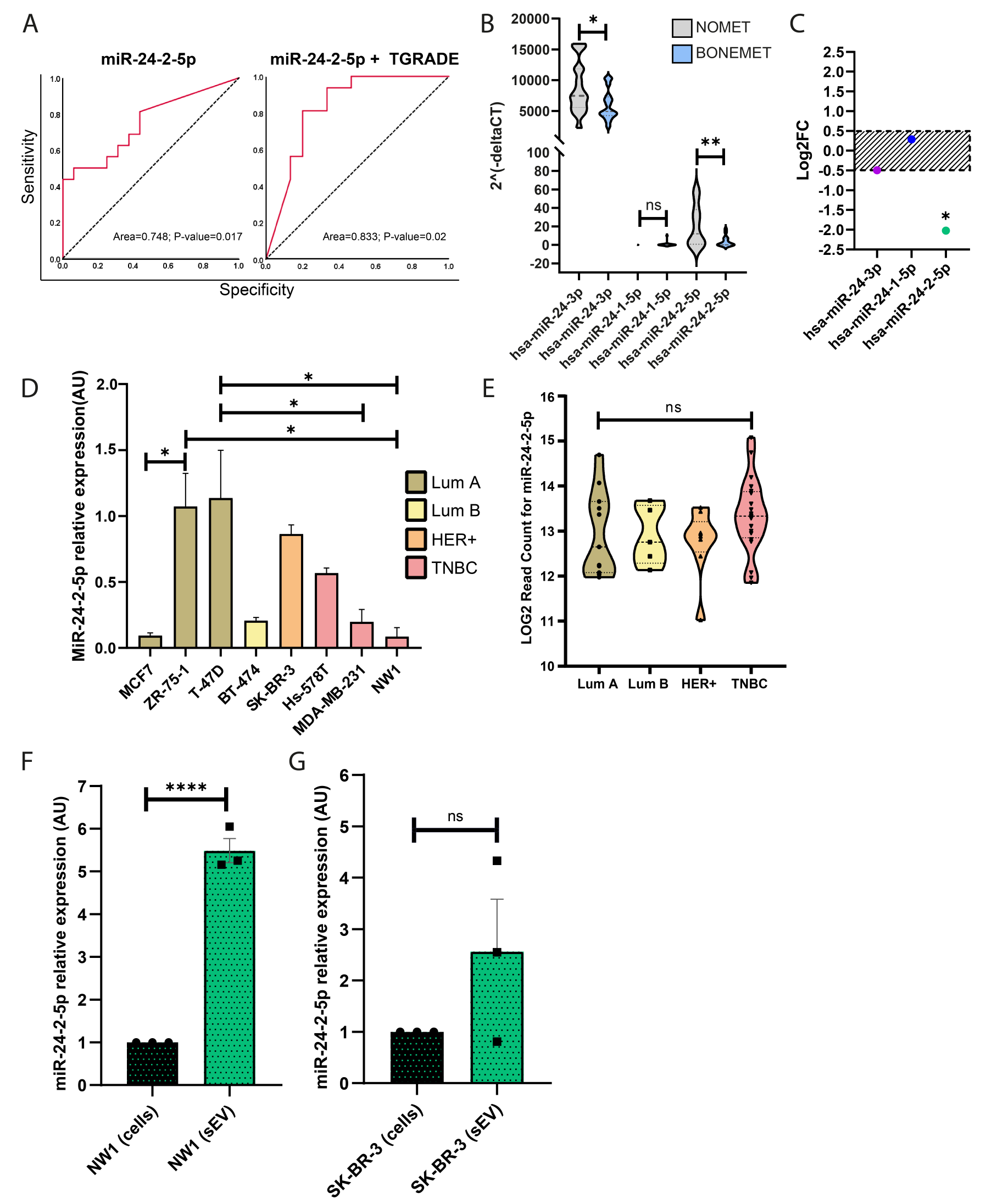
The expression levels for miR-24 forms in serum samples from 48 early-stage BC patients were obtained from our published dataset [10], where a miRome screen was conducted by TaqMan low-density arrays (TLDA). In this dataset, patients have been divided into 3 groups based on clinical information on patients’ metastatic status: NOMET (n = 16) with no metastatic recurrence, BONEMET (n = 16) with a first relapse in bone, and SOFTMET (n = 16) with a first relapse in other tissues than bone [10]. Clinical details on BC patients’ cohort can be found in supplementary Fig. S1. Raw cycle threshold (CT) values, normalized delta CTs (ΔCT) (by global mean normalization), and expression levels for miR-24-1-5p, miR-24-2-5p, miR-24-3p were retrieved from this dataset as well as data on the differential expression of these miRNAs between NOMET and BONEMET groups.
Receiver operating characteristic (ROC) curves analysis
Combined ROC curve analysis was constructed to evaluate the value of circulating miR-24-2-5p levels combined with tumour grade patients’ information in distinguishing between NOMET and BONEMET groups. Areas under the ROC curves (AUC) were calculated based on 2−ΔCT values.
Cell lines and cell culture
MDA-MB-231, T-47D, MCF7, BT-474, ZR-75-1, SK-BR-3, and Hs-578T human breast cancer lines were obtained from the American Type Culture Collection (ATCC) and authenticated in-house by DNA fingerprinting, using short tandem repeat method of 10 loci. MDA-MB-231-luc2-NW1 (NW1) cell line was obtained from Dr Ning Wang, University of Sheffield, Sheffield, UK [16]. NW1/LENTI-Ctrl-GFP + were generated by lentivirus transduction as previously described [10]. MDA-MB-231, NW1, T-47D, MCF7, Hs-578T, and BT-474 cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose (GlutaMAX, Gibco, UK), ZR-75-1 in RPMI 1640 medium (Gibco, UK), SK-BR-3 in McCoy’s 5 A medium (Sigma), supplemented with 10% (v/v) FCS (Invitrogen) and 100 U/mL penicillin/streptomycin, at 37 °C, 5% CO2. Cell cultures were routinely tested for mycoplasma contamination (MycoAlert PLUS Mycoplasma Detection Kit, Lonza). Cell lines in culture were used with a maximum of 20 passages after receipt.
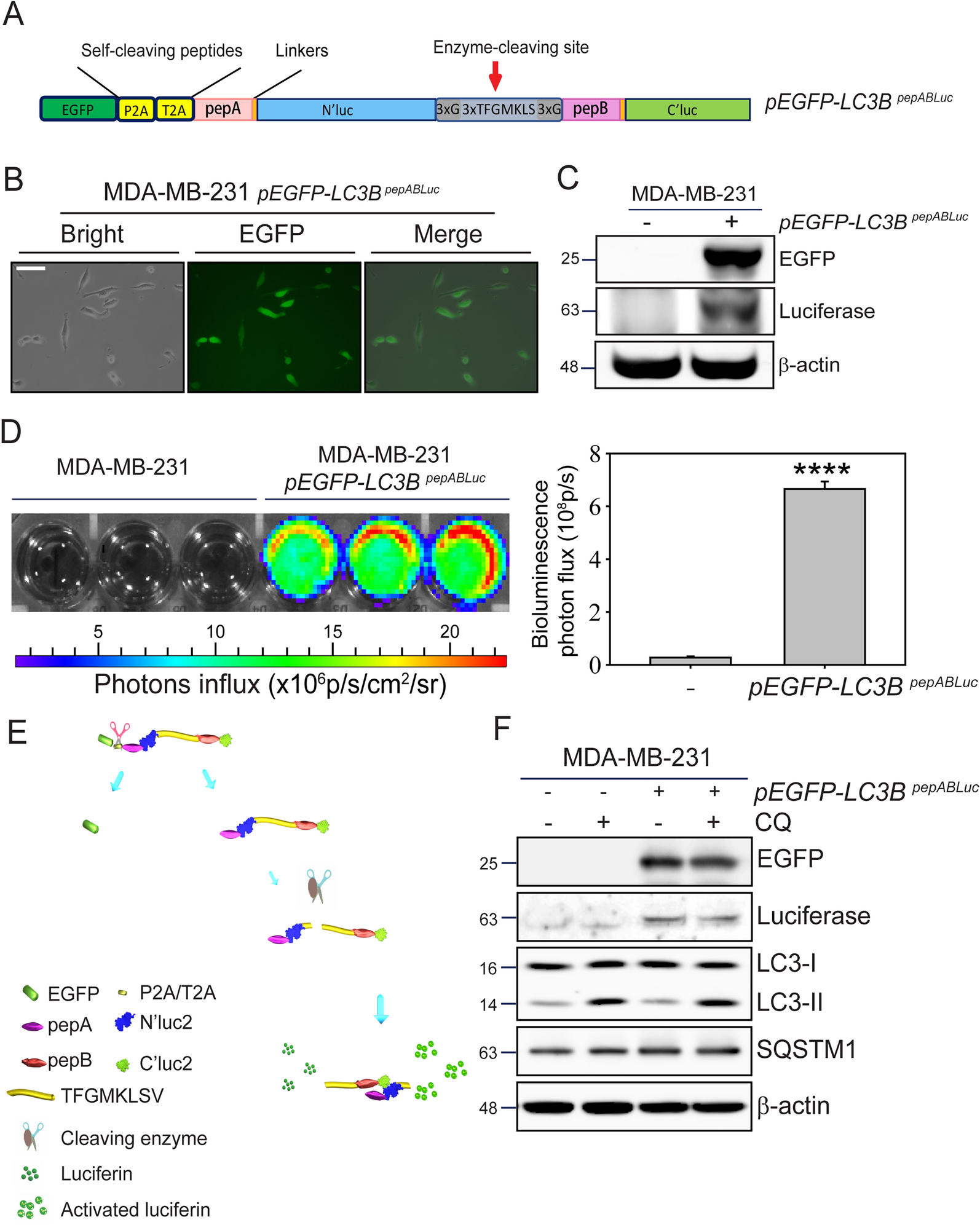
MiR-24-2-5p transient and stable overexpression in BC cells
Transient miR-24-2-5p overexpression in BC cell lines (NW1/MIMIC-miR-24-2-5p, MCF7/MIMIC-miR-24-2-5p) was obtained by cell transfection with Lipofectamine2000 (Invitrogen) using 50 nM of miR-24-2-5p mimics (GenePharma, Shanghai). Control cells (NW1/MIMIC-negCTRL, MCF7/MIMIC-negCTRL) were transfected with 50 nM FAM-labelled negative control mimics (GenePharma, Shanghai). Transfected cells were used for in vitro and in vivo experiments at least 24 h after transfection.
Stable miR-24-2-5p cell lines (NW1/miR-24-5p, MCF/miR-24-5p) were generated by lentivirus transduction with a shMIMIC human lentiviral microRNA hsa-miR-24-2-5p hCMV-TurboGFP (Horizon Discovery, United Kingdom) at 10 MOI, and clones were selected for 2 weeks with 8 µg/mL puromycin. Control cell line for NW1 and MCF7 were obtained by transduction of SMARTvector non-targeting hCMV-TurboGFP control particles as previously described [10].
MiR-24-2-5p transient downregulation in BC cells
Transient miR-24-2-5p downregulation in BC cell lines (ZR-75-1/INHIB-miR-24-2-5p, T-47D/INHIB-miR-24-2-5p, SK-BR-3/INHIB-miR-24-2-5p) was obtained by cell transfection with Lipofectamine2000 (Invitrogen) using 50 nM of hsa-miR-24-2-5p mirVana miRNA inhibitor (Ambion, Thermo Fisher). Control cells (ZR-75-1/INHIB-negCTRL, T-47D/INHIB-negCTRL, SK-BR-3/INHIB-negCTRL) were transfected with 50 nM mirVana miRNA inhibitor Negative Control #1 (Ambion, Thermo Fisher). Transfected cells were used for in vitro experiments at least 24 h after transfection.
MiR-24-2-5p quantification in BC cells
Mir-24-2-5p expression levels were quantified by TaqMan real-time quantitative PCR (RT-qPCR) following manufacturers’ instructions. Total RNA was extracted with miRNeasy kit (Qiagen), following manufacturers’ instructions. RNA concentration and purity were evaluated using a Nanodrop™ 2000 spectrophotometer (Thermo Scientific). A reverse transcription into cDNA was performed with TaqMan microRNA RT kit and specific probes directed to hsa-U6 and hsa-miR-24-2-5p (TaqMan miRNA Assays). Real-time qPCR was performed with TaqMan Universal Master Mix NO UNG. Relative gene expression was calculated using the 2−ΔΔCT method.
Quantification of transcript expression levels in BC cells and osteoclasts
RNA from cell pellet was extracted as described above. Reverse transcriptions into cDNA were performed with iScript cDNA Synthesis Kit (Bio-Rad), and real-time qPCR was carried out with SsoAdvance Universal SYBR Green Supermix (Bio-Rad), following manufacturers’ instructions. Relative gene expression was calculated using the 2−ΔΔCT method. Primer sequences are listed in supplementary Table S1 or were previously published [17].
Small extracellular vesicle (sEV) isolation by ultracentrifugation
NW1 cells were seeded at a low percentage of confluency (20–30%) and cultured in complete medium for 24 h. The complete medium was then replaced with sEV-free medium (sEV-deprived medium by ultra-centrifugation). After 72 h in culture, the conditioned medium was collected, and sequential centrifugations at 4 °C were performed (300*g for 10 min, 10.000*g for 10 min, 100.000*g for 70 min). An Optima XPN-80 Beckman Coulter Ultracentrifuge was used for the ultra-centrifugation steps. sEV pellet was resuspended in QIAzol (Qiagen) for downstream RNA analysis.
Proliferation assay
Twenty-four hours post-transfection, cells were seeded into 12-well plates (Costar), in triplicate, at a concentration of 1 × 104 cells/500 µL/well. For 5 consecutive days (day 0, 1, 2, 3, 4), cells were washed with PBS, fixed with 4% (v/v) paraformaldehyde (Fisher Scientific, UK), and stained with crystal violet solution (Sigma-Aldrich). The optical density (590 nm) of a solubilized crystal violet solution in 10% (v/v) acetic acid was measured using a SpectraMax M5e microplate reader (Molecular Devices), and readings were normalized to the measurement obtained at day 0.
Transwell migration and invasion assays
Twenty-four hours post-transfection, cells cultured at 80% confluence were treated with 10 µg/mL of mitomycin C (Sigma) to prevent proliferation prior performing the cell migration assay. Tumour cells were then resuspended in serum-free DMEM medium and seeded at a concentration of 104 cells/200 µL/well in the upper chamber of a 24-well plate (Costar), while complete DMEM medium was used as a chemoattractant in the lower chamber of the well. After 24 h, cells in the upper chamber were removed with a cotton swab, and cells that had migrated through the porous membrane (8-µm diameter pore-size) were fixed in 100% ethanol, H&E stained, imaged under a Leica RMRB automatic upright microscope, and analysed with ImageJ 1.53k, Java 1.8.9_172 (64-bit) software. Tumour cell invasion experiments were conducted in 24-well plates in the same way as cell migration assays except 8-µm diameter pore-size polyethylene terephthalate (PET) membrane inserts were coated with Matrigel® (BioCoat, Costar).
Conditioned medium (CM) preparation from BC cells and mir-24-2-5p quantification
Six hours post-transfection, culture media of MIMIC-transfected NW1 (NW1/MIMIC-miR-24-2-5p, NW1/MIMIC-NegCtrl) and MCF7 (MCF7/MIMIC-miR-24-2-5p, MCF7/MIMIC-NegCtrl) BC cells were replaced with serum-free DMEM medium. BC cell-conditioned media (CM) were then collected after 24 h, centrifuged to remove cell debris, aliquoted, and stored at -80 °C until use for the osteoclastogenesis assay. Total RNA was extracted from 200uL of CM by using a NucleoSpin® miRNA Plasm (Macherey-Nagel kit, Germany), accordingly to manufacturer instructions. MiR-24-2-5p expression levels were quantified as described above.
Osteoclastogenesis assay
Osteoclastogenesis assay with human and murine monocytes was performed as previously described [10].
Briefly, primary human osteoclasts were differentiated from human peripheral blood mononuclear cells of healthy donors. The procedure was approved by the Ethical Committee of Campus Bio-Medico University of Rome (Prot. 21/15 OSS), and in accordance with the Declaration of Helsinki principles. Briefly, 1*104 CD14 + monocytes resuspended in complete RPMI medium supplemented with M-CSF (25 ng/mL) and RANKL (50 ng/mL) were seeded in 96-well plates. After 72 h, cell culture medium was replaced with complete medium supplemented with M-CSF and RANKL, with or without BC cell-CM (1:16 volume dilution). Medium was replaced every 3 days. At day 12, cells were fixed and stained for TRAP activity (Sigma), or cell pellet was collected for gene expression analysis. Mature multinucleated osteoclasts (> 3 nuclei) were enumerated, and the total area covered by osteoclasts measured using a Nikon NIS-Elements microscope imaging software.
For murine osteoclastogenesis, bone marrow cells from tibiae and femora of 6/8-week-old OF1 male mice (Charles River) were flushed, centrifuged, resuspended in Ficoll® Paque Plus (Cytiva), and further centrifuged to allow the isolation and enrichment of mononuclear cells. 1*10⁵ cells from the isolated mononuclear cell fraction were then seeded in 12-well plate wells, and cultured 24 h in α-MEM medium containing 10% (v/v) FCS (Invitrogen) with M-CSF (20 ng/mL). The next day, culture medium was replaced with a differentiation MEM-α medium containing 10% (v/v) FCS, M-CSF (20 ng/mL), and RANKL (10 ng/mL). For experiments conducted with BC cell-CM, mononuclear cells were continuously exposed to the CM (1:16 volume dilution), and mature multinucleated osteoclasts were then fixed after 7 days, stained for TRAP activity, and counted, or cell pellet was collected for gene expression analysis. For osteoclastogenesis experiments designed to measure endogenous miR-24-2-5p levels during the differentiation into osteoclasts of bone marrow-derived mononuclear cells treated with M-CSF and RANKL, bone marrow cells were fixed, TRAP stained, and counted at early and late time points, which correspond to immature and mature osteoclasts, respectively. Alternatively, cell pellets were collected at these early and late stages of osteoclast differentiation for gene expression analysis.
TargetScan target prediction and ClueGO-based analysis
TargetScanHuman 7.0 software (https://www.targetscan.org/vert_70/, Accessed April 2021) was used to predict miR-24-2-5p targets in human transcriptome by searching for the presence of 8mer, 7mer, and 6mer sites that matched with miR-24-2-5p seed region. Top 250 predicted targets (arbitrary threshold) were used to perform a ClueGO-based analysis [18] using the ‘GO Biological Process’ database.
RNA-seq of MIMIC-transfected BC cells
Thirty-six hours post-transfection, cell pellets from MIMIC-transfected NW1 (NW1/MIMIC-miR-24-2-5p, NW1/MIMIC-NegCtrl) or MCF7 (MCF7/MIMIC-miR-24-2-5p, MCF7/MIMIC-NegCtrl) BC cells from three independent experiments were collected and stored at -80 °C until use.
Total RNA was extracted using the RNeasy Mini Kit (Qiagen), according to the manufacturer’s recommendations. A DNase digestion step was performed during the RNA extraction using RNase-free DNase I Kit (Qiagen). RNA quantity and purity were evaluated using a 2100 Bioanalyzer (Agilent) and a Qubit RNA IQ Assay (ThermoFisher). RNA extracts (675 ng) were then used for RNA-seq library preparation with Poly-A enrichment.
A single-end RNA-seq was undertaken on the Illumina HiSeq™ 2500 platform in rapid run mode using the Illumina HiSeq™ Rapid Cluster Kit (Illumina, Inc., San Diego, CA, USA). Each sample had 15 million reads. RNA-seq data was aligned to GRCh38 human genome assembly using STAR v2.7.5c, and transcript quantification was performed using RSEM v1.3.1 as previously described [10]. Poorly expressed transcripts (< 0.5 counts per million in all samples) were eliminated for further analysis. Counts were normalized by weighted trimmed mean of M-values using TMM function of EdgeR Bioconductor package as previously described [10].
Differential gene expression (DE) analysis and gene set enrichment analysis (GSEA)
DE analysis, based on negative binomial generalized linear models, was performed using EdgeR Bioconductor package to compare MIMIC-miRNA-24-2-5p transfected cells to MIMIC-negCTRL transfected cells [10].
GSEA analysis was performed using ClusterProfiler Bioconductor package on genes ranked by the fold change estimated in DE analysis. A manual annotation of gene networks has been performed based on the description of each gene network retrieved from GSEA website (https://www.gsea-msigdb.org/gsea/index.jsp, Accessed May 2022).
Animal studies
Animal experiments conducted at the University of Sheffield (Sheffield, UK) were performed using 6-to-7-week-old female BALB/c fox/- nude mice (Charles River, Kent, UK) kept on a 12-h/12-h light/dark cycle with free access to food and water. NW1 cells transfected with MIMIC-miR-24-2-5p or negative control cells were injected into the left cardiac ventricle (5 × 104 cells) of mice 72 h post-transfection. Tumour growth was monitored in anesthetized mice using an IVIS Lumina II system (Caliper Life Sciences, UK), following subcutaneous injection of D-Luciferin (Invitrogen, UK) to animals 4 minutes before imaging. Total blood was collected by cardiac puncture on the sacrifice day (9 days after intracardiac injection of tumour cells), aliquoted, and stored at -80 °C for downstream analysis by ELISA. An additional ex vivo imaging of hindlimbs and major organs by IVIS was performed at the autopsy to confirm site of metastases.
Enzyme-linked immunosorbent assays (ELISA)
Serum concentrations of CTX-I (C-terminal telopeptide of type I collagen) and P1NP (pro-collagen type I N propeptide) were measured by ELISA using commercially available kits (E-EL-M3023 and E-EL-M3033, Elabscience), following manufacturers’ instructions.
Statistical analysis
For combined ROC curve analysis (miR-24-5-5p with tumour grade), statistical significance was set at p < 0.05 (two-sided test), and a binary logistic regression was calculated using IBM SPSS Statistics. Broad institute software Morpheus was used to generate the plot. Survival analysis was performed by dividing patients into two groups [high (value > 0.1) and low (value <-0.1)] based on miR-24-2-5p expression (Z-score of 2−ΔCT). The Kaplan–Meier estimator was used to determine the relationship between miRNA expression and patient distant recurrence-free survival (only in bone). Differences in survival across the strata were calculated using a log-rank p-test.
All statistical analyses on experimental data were performed using Prism GraphPad 9.2.0 (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was measured using parametric testing (Student’s t-test), assuming equal variance, and defined as p value (p) ≤ 0.05. All graphs represent mean ± standard error mean (SEM), *p ≤ 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
All in vitro experiments consisted of at least 3 independent biological repeats with appropriate controls.
For in vivo experiments, power calculations for animal experiments are based on our previous work [10, 19, 20]. In experiments conducted with NW1 cells, 70–80% of animals have metastasis. Assuming a power of 80% and a level of significance of 5%, we estimated that we will be able to measure a difference of 60% or greater with 9 animals per group, using a Mann-Whitney test.
For Cancer cell line Encyclopaedia (CCLE) analysis, information of miRNA-24-2-5p expression levels in 44 BC cell lines was retrieved from the public database. The violin plot was used to visualise the distribution of miRNA expression levels, which were reported as log2 read counts. Statistical significance was evaluated using the unpaired t-test, where p-values 0 < 0.05 were considered as significant.
For the TCGA, selected matched (RNA-seq and small RNA-seq) data sets from 736 patients were analysed. Pearson correlation was conducted to identify the association between miR-24-2-5p (RPM values, Z-score across the 736 samples) expression and target mRNA expression (TPM values, Z-score across the 736 samples).




留言 (0)