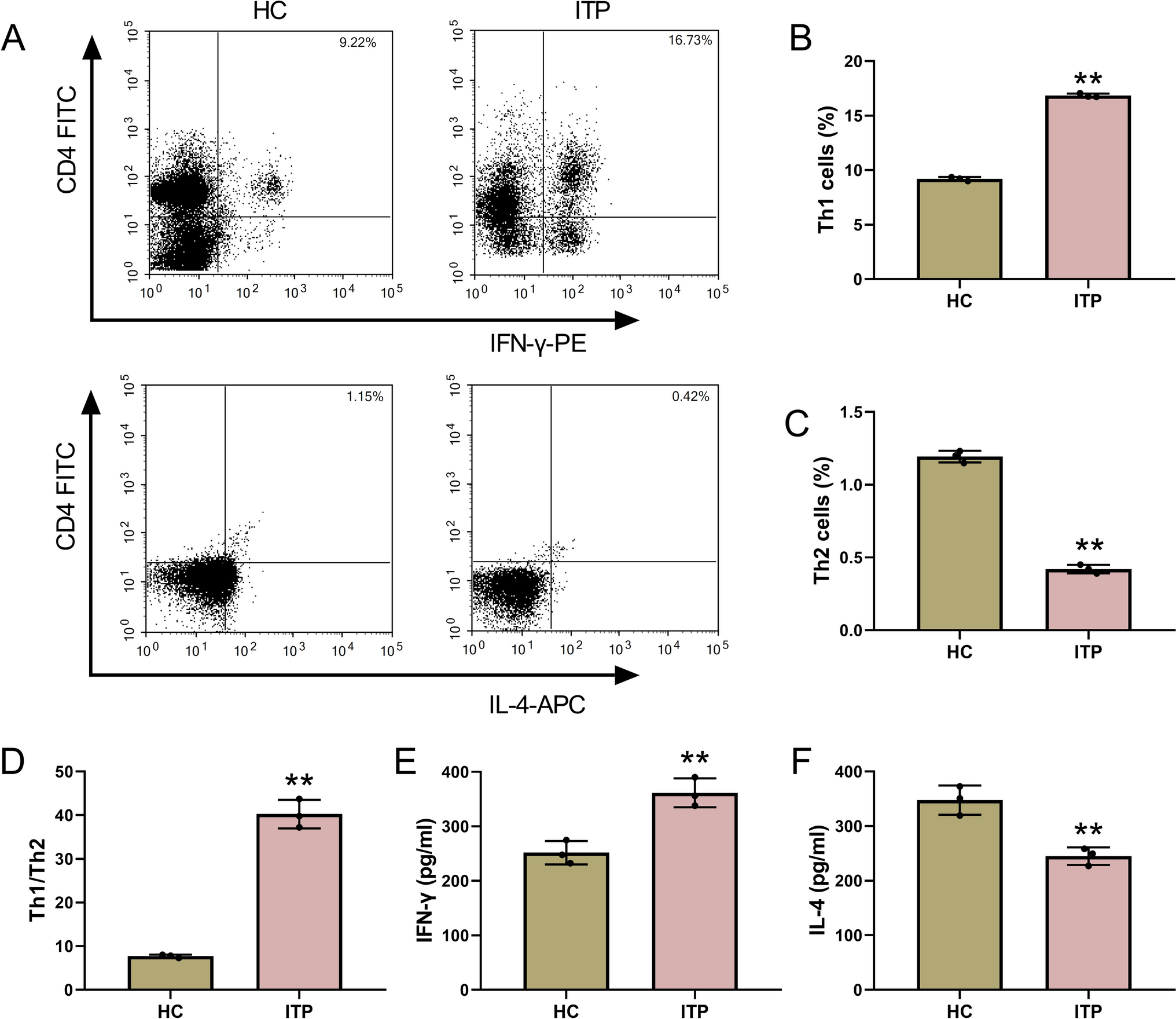
Data acquisition in GEO and public-access databases
We downloaded and used processed data from the GSE171023, and GSE231835 datasets deposited in the GEO (https://www.ncbi.nlm.nih.gov/geo/). The GSE171023 dataset contains m(6)A-sequencing data for bladder cancer T24 cells with IGF2BP3 knockout vs. control. The GSE231835 dataset contains RNA-sequencing data for bladder cancer T24 cells with cisplatin resistance vs. control. The public-access database, UALCAN (https://ualcan.path.uab.edu/index.html), was searched to show candidate gene expression based on the TCGA samples of bladder cancer. The RBP-Target analysis using the ENCORI platform (https://rnasysu.com/encori/index.php) and the m6A target database (http://m6a2target.canceromics.org/) were searched to predict the possible sites of m6A modification of SRD5A3.
Human tissue specimen selection
Primary tumor samples and adjacent normal samples from 30 bladder cancer patients who had undergone radical or partial cystectomy were collected for clinical validation. None of included patients received chemotherapy or radiotherapy before surgical treatment. Among these 30 bladder cancer patients, there were 21 males and 9 females. Based on tumor node metastasis (TNM) staging system detailed in the American Joint Committee on Cancer [28], 30 bladder cancer patients consisted of 22 with stage I or stage II and 8 with stage III, 26 being well differentiated and 4 being poorly differentiated, and 2 with lymph-node metastasis and 28 without. Human tissue specimen selection was performed with the approval of the Ethics Committee of Guangzhou Institute of Cancer Research, the Affiliated Cancer Hospital, Guangzhou Medical University (No. 2024-ZN032).
Cell culture and lentiviral transduction
Human bladder cancer cell lines, T24 and 5637 (ATCC, Shanghai, China), were cultured in the RPMI-1640 medium (Gibco, Gaithersburg, MD, USA) with the addition of 10% fetal bovine serum (FBS, Gibco). Normal human uroepithelial cell line, SV-HUC-1 (Cell Bank of Type Culture Collection, Shanghai, China), was cultured in the DMEM (Invitrogen, Carlsbad, CA, USA) with the addition of 10% FBS (Gibco). All cells underwent incubation in a humidified atmosphere containing 5%CO2 and 95% at 37 °C. Parental T24 and 5637 cells were exposed to gradually increasing doses of cisplatin over the course of 6 months to develop cisplatin-induced drug-resistant bladder cancer cell lines, namely T24R and 5637R cells. At the beginning, T24 and 5637 cells were exposed to a sub-lethal dose of cisplatin which was set just below the level inducing significant cell death while ensuring the survival of a small population of less sensitive cells. To generate the stable cell line, SRD5A3 knockdown (KD) virus, IGF2BP3 KD virus, IGF2BP3 overexpression (OE) virus, and their counterpart control virus were purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China). The lentiviral transduction was preformed based on the manufacturer’s instructions. Supernatants of 293 T cells infected by these lentiviruses were harvested 48 h later followed by purification using ultracentrifugation. T24R and 5637R cells were transduced with the recombinant lentivirus plus 5 μg/ml of Polybrene. A stable transfected strain was selected by a medium with 4 μg/ml puromycin for two generations.
Quantitative real-time PCR (qRT-PCR)
Extraction of total RNA from human tumor tissue and bladder cancer cells and the following reverse transcription into cDNA were completed by using Trizol reagent (Invitrogen) and PrimeScript RT reagent (Takara, Tokyo, Japan), respectively. The mRNA expression levels of interest genes were determined using SYBR Premix Ex Taq (Takara) with the aid of a Bio-Rad CFX96 PCR system (Bio-Rad, Hercules, CA, USA). The resulting mRNA expression levels of interest genes were normalized GAPDH mRNA and quantification of their relative expressions was completed by the 2−∆∆Ct method. Used primer sequences are: 5’-TTTAATCAGGCCCTGTCTGC-3’ (forward) and 5’-GGGGTATAGAAATGGAATGGAGA-3’ (reverse) for SRD5A3, 5’-TCGTGACCAGACACCTGATGAG-3’ (forward) and 5’-GGTGCTGCTTTACCTGAGTCAG-3’ (reverse) for IGF2BP3, 5’- GGAGCGAGATCCCTCCAAAAT-3’ (forward) and 5’-GGCTGTTGTCATACTTCTCATGG-3’ (reverse) for GAPDH.
mRNA stability assay
T24R cells and 5637R cells (control and IGF2BP3 knockdown) were seeded into 6-well plates and then treated with 5 µM actinomycin D (Sigma, Missouri, USA) for indicated time points (0, 2, 4, 6, 8, and 10 h), followed by qRT-PCR. The level of remaining mRNA at 2, 4, 6, 8, and 10 h was normalized to the level at 0 h. The one-phase exponential decay curves (from 0 to 10 h) were plotted to reflect mRNA decay kinetics.
Immunoblotting analysis
The human tumor tissue homogenates and bladder cancer cells reacted in the RIPA lysis buffer to obtain total protein, followed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (10%) separation, transfer onto polyvinylidene fluoride membrane, and blocking buffer treatment (5% skim milk in TBST). The membranes were incubated with rabbit polyclonal anti-SRD5A3 antibody (PA5-55480, Invitrogen, Carlsbad, CA, USA), mouse monoclonal anti-IGF2BP3 antibody (MA5-27480, Invitrogen), and mouse monoclonal anti-GAPDH antibody (MA5-15738, Invitrogen), followed by rinse with TBST and incubation with secondary antibodies. The signal of immunoblots was visualized using an ECL chromogenic kit (Beyotime, Shanghai, China) and GAPDH normalized the density of each immunoblot. Densitometry analysis of immunoblots was carried out with the aid of the ImageJ software program (NIH, Bethesda, MA).
CCK-8 assay
T24R and 5637R cells were seeded in 96-well plates (5000 cells in 100 μL per well) in a complete medium and each well was added with 100 μL CCK-8 solution (10% of the total volume) (10 µL, Transgen, Beijing, China) for 2 h incubation. The absorbance at 450 nm was captured. Then, the medium was replaced by complete medium supplemented with 5 μg/ml CDDP, and the plates were incubated for 24 h 48, and 72 h. After each interval, each well was added with CCK-8 solution for 2 h incubation, with the absorbance at 450 nm read.
Colony formation assay
T24R and 5637R cells were seeded in 6-well plates (500 cells per well) and treated with 5 μg/ml CDDP for 24 h. Then, the cells were maintained in DMEM containing 10% FBS for 2 weeks. The medium was refreshed every two days. After fixing with 4% paraformaldehyde (24 ℃ for 1 h) and staining with 0.1% crystal violet (30 min), colonies with more than 50 cells were imaged and counted for statistical analysis.
EdU staining
The BeyoClick™ EdU Cell Proliferation Kit (Beyotime) was employed to further examine cell proliferation. T24R and 5637R cells were seeded in 96-well plates (1000 cells per well) and treated with 5 μg/ml CDDP for 24 h. Each well was added with 150 μL EdU solution. After 2 h, each well was added with 4% paraformaldehyde for 30 min. Following 5 min permeabilization, the cells were treated with 50 μL EdU-Click reaction mix for 30 min. The cell nuclei were counterstained with 1 mg/ml HO33342 (Beyotime), and the stained cells were captured under a fluorescence microscope (Nikon, Japan) and imaged.
Flow cytometry
The Annexin V-FITC/PI Apoptosis Detection Kit (BD Biosciences, San Deigo, CA, USA) with the aid of the BD FACSCalibur flow cytometer (BD Biosciences) was utilized for cell apoptosis assay. T24R and 5637R cells were seeded in 6-well plates (1000 cells per well) and treated with 5 μg/ml CDDP for 24 h. Then, the cells were resuspended in 300 ml of binding buffer followed by the addition of Annexin V-fluorescein isothiocyanate and propidium iodide, 5 μL for each stain.
Methylated RNA immunoprecipitation-PCR (MeRIP-qPCR)
The Magna MeRIP™ m6A kit (Merck Millipore, MA, USA) was utilized to perform MeRIP procedure. In brief, total RNA from cells were purified by an mRNA Isolation System (Sigma, Missouri, USA), treated by DNase I for 30 min, and made into random fragments (100 nucleotides). Subsequently, the RNA fragments were incubated with a mixture of protein-A/G magnetic beads and m6A antibody (MA5-33030, Invitrogen) or IgG (MA5-42729, Invitrogen). The co-precipitated RNAs were separated using the elution buffer followed by the qRT-PCR procedures to determine the m6A methylation level of SRD5A3.
RNA immunoprecipitation (RIP) assay
The Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore) was utilized to perform RIP assay of IGF2BP3 and SRD5A3. After treating with a complete RIP lysis buffer (Millipore) in the presence of protease inhibitors and RNase, cell lysates were incubated with either anti-IGF2BP3 antibody (712139, Invitrogen) or IgG (MA5-42729, Invitrogen) and immunoprecipitated with magnetic Protein A/G beads. After proteinase k treatment, the immunoprecipitated RNA and total RNA from the whole cell lysates (input controls) were separated using the elution buffer followed by the qRT-PCR procedures to determine the mRNA enrichment of SRD5A3.
m6A mutation and luciferase reporter assay
The SRD5A3 3ʹUTR fragments which contained intact m6A sites and mutant m6A motifs (mutant 1, 2, 3, and 1 + 2 + 3), were inserted into the pGL3-control vectors (Promega, Madison, WI, USA). Marked adenosine (A) was replaced by cytosine (C) to generate mutant m6A motifs. For dual-luciferase reporter assays, 100 ng wild-type or mutant SRD5A3 fragments, 200 ng shRNA against IGF2BP3 or scramble shRNA, and 20 ng pRL-TK (Renilla luciferase control reporter vector) were co-transfected into HEK-293 T cells in 24-well plates and incubated for 24–36 h. The Dual-Glo Luciferase system (Promega) was employed to examine the luciferase activity using and the pRL-TK activity normalized the results.
Animal experiments
Forty Male BALB/c 5–6 weeks nude mice (Charles River, Beijing, China) were implanted subcutaneously with 1 × 106 transfected T24R cells (NC, SRD5A3-KD, NC, and IGF2BP3-KD) or untransfected T24R cells coated with Matrix (Becton Dickinson, Bedford, MA, USA). Once the average volume of subcutaneous tumors had grown to be approximately 100 mm3, the mice implanted subcutaneously with untransfected T24R cells were injected with recombinant lentivirus (NC, SRD5A3-KD, NC, and IGF2BP3-KD) via the tail vein [the final lentiviral vector titers were determined to contain 3 × 108 TU per mouse, followed by intraperitoneal injections of CDDP (2 mg/kg) every 3 days for 35 days. On day 35, the mice were euthanized by exposure to prolonged inhalatory anesthesia, and their xenografted tumors were excised, photographed, and measured for weight. All procedures involving experimental animals were approved by Guangzhou Medical University (No. GY2024-572). Animals were treated in strict accordance with the Guidelines for the Care and Use of Laboratory Animals (Eighth Edition) [29]. Significant efforts were done to minimize both animal suffering and the numbers used.
Statistical analysis
Six independent experiments yielded results presented as mean with standard deviation (s.d.). All statistical analyses involving Student’s t test, one-way analysis of variance (ANOVA) plus Tukey’s post hoc test, and repeated-measures ANOVA plus Sidak’s multiple comparisons test were performed with the aid of GraphPad Prism version 8.0 (GraphPad Software, La Jolla, CA, USA) for Windows. A value of P < 0.05 was regarded statistically significant.





留言 (0)