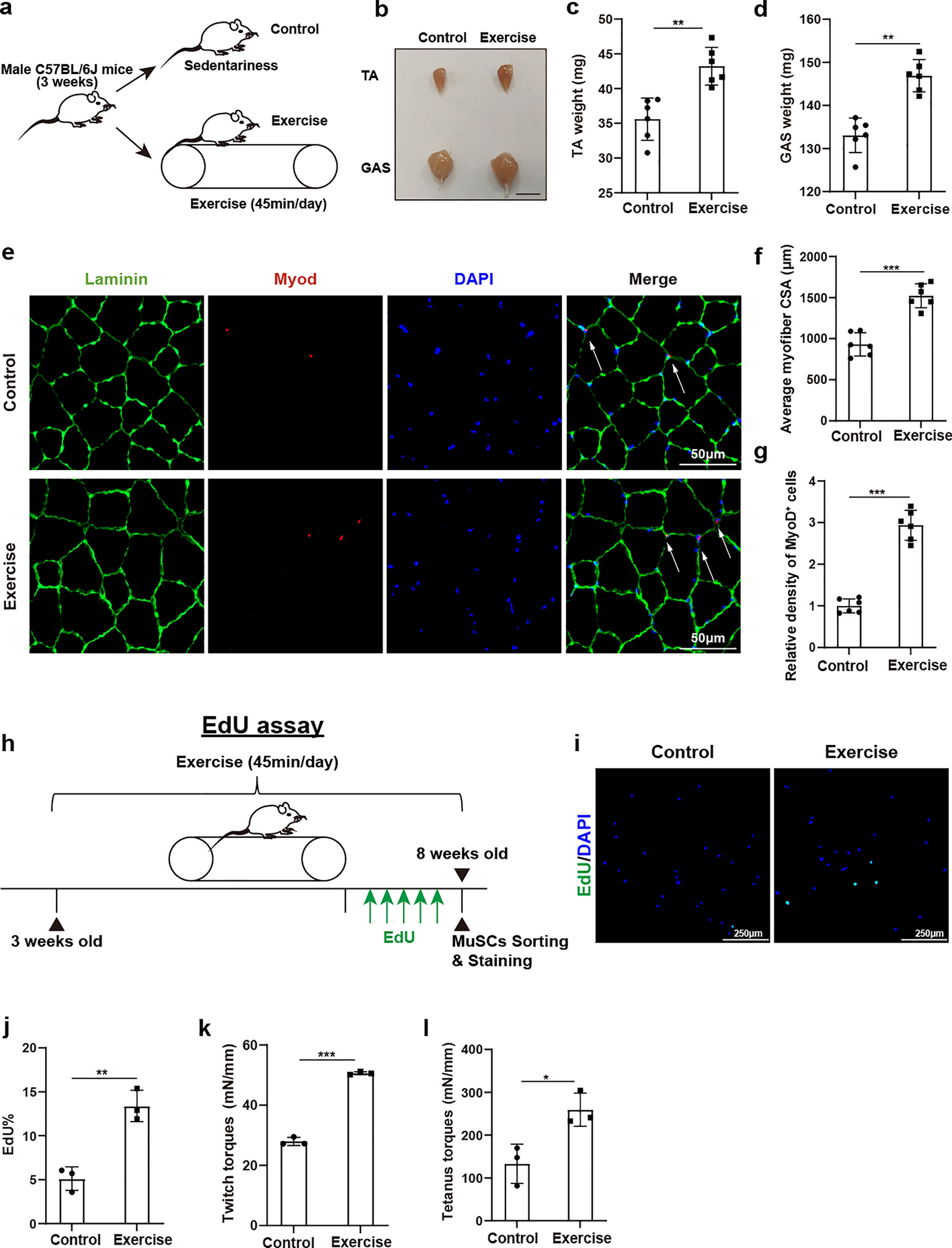
Animals and exercise model
Animal experimentation protocols were approved by the local institution’s ethical committee (approval no. XHEC-F-2024-042). Wild-type C57BL/6J male mice aged 3 weeks were purchased from GemPharmatech (Nanjing, China). Pax7-CreERT2 mice (011763) were procured from Jackson Laboratory, while Mettl3f/f mice (strain no. T006659) were obtained from GemPharmatech (Nanjing, China). MuSC-specific Mettl3 conditional knockout mice were induced through intraperitoneal administration of 100 µl of 10 mg/ml tamoxifen every 2 days from 2 to 3 weeks of age. The mice were allowed ad libitum access to food and water under a 12-h light/dark cycle. The exercise model was established in accordance with a modified protocol as described previously [24, 25]. Briefly, mice underwent a familiarization phase on the treadmill for 5 days, and the speed was gradually increased from 8 m/min to 12 m/min, for 30 min daily. Subsequently, the mice were subjected to 4 weeks of treadmill training at a speed of 12 m/min for 45 min/day, 5 days/week.
Isolation of MuSCs
The lower-limb skeletal muscles were obtained and digested following established methodologies [26,27,28]. Briefly, the fresh skeletal muscle was first finely minced to a meat-paste consistency, digested for 1 h using collagenase II (Worthington Biochemical, 700–800 U/mL), followed by an additional 30 min with a mixture of collagenase II and dispase (11 U/mL). The resultant mixture underwent ten passes through a 20 gauge needle, followed by filtration through a 40 µm cell strainer (Corning). To isolate MuSCs, the cell suspension was incubated with following antibodies for 45 min at 4 °C: a cocktail of APC anti-CD45, APC anti-CD31, Biotin anti-VCAM1, and FITC anti-Sca1. Subsequently, PE/Cy7 streptavidin was used for staining with an additional 15 min. Finally, using a BD Influx sorter, the CD31−/CD45−/Scal1−/VCAM1+ MuSCs were isolated by fluorescence-activated cell sorting (FACS).
EdU assay
During the final week of the exercise experiment, EdU (Yeasen, 40284ES76, 5 mg/kg body weight) was administered intraperitoneally daily to mice. Twelve hours later, MuSCs were isolated using FACS. The freshly isolated MuSCs were subsequently plated and fixed in 4% paraformaldehyde for further experiments.
Cell culture and differentiation
In collagen-coated dishes, primary MuSCs were cultured in F10 basal medium supplemented with 20% FBS, 5 ng/mL IL-13, 5 ng/mL TNF-α, 5 ng/mL IFN-γ, 5 ng/mL IL-1α, 2.5 ng/mL bFGF, and 1% penicillin–streptomycin under optimal conditions of 37 ℃ with 5% CO2, in line with previous literature [26]. The differentiation medium utilized was Dulbecco’s modified Eagle medium (DMEM) supplemented with 2% horse serum and 1% penicillin–streptomycin.
Immunofluorescence staining
Cryosections or cultured cells were fixed in PBS containing 4% paraformaldehyde (Merck, 158,127) for 15 min, permeabilized in 0.5% Triton X-100 for 15 min at room temperature, and then blocked with 1% BSA (Sigma-Aldrich, 9048–46-8) in PBS for 1 h. Subsequently, the samples were incubated overnight at 4 °C with anti-Pax7 (Thermo Fisher Scientific, PA1-117, 1:100), anti-MyoD (Santa Cruz Biotechnology, sc377460, 1:200), anti-MyoD (Santa Cruz Biotechnology, sc760, 1:200), anti-MyoG ( Santa Cruz Biotechnology, sc-12732, 1:200), anti-Laminin (Abcam, ab11575, 1:500), anti-Mettl3 (Abcam, ab195352, 1:1000), and anti-MyHC (Thermo Fisher Scientific, MA5-35613, 1:1000). Following this, secondary antibodies were used to incubate the samples for 1 h. Nuclei were subsequently stained with DAPI (Sigma-Aldrich, S7113), and antifade mounting media (Invitrogen, p36935) was applied to cover the samples. Finally, a Leica SP8 confocal microscope was employed to capture the images. Immunofluorescence staining for Laminin was used to determine the boundaries of myofibres. Immunofluorescence staining of MyHC was conducted to identify the outline of myotubes. For each sample, a minimum of five independent fields of view were randomly selected for evaluation. ImageJ was applied to quantify the cross-sectional area (CSA) of myofibers and the diameter of myotubes.
Gene expression analysis
The EZ-press RNA Purification kit (EZBioscience) was used to extract total RNA following the manufacturer’s instructions. Reverse transcription was then performed using MuLV reverse transcriptase (NEB, M0253S). Subsequently, the products were subjected to quantitative PCR reactions with Genious 2× SYBR Green Fast qPCR mix (Abclonal, RK21205) in the CFX96 real-time PCR system. The primers used are listed in Additional file 1: Table S1.
Overexpression
Briefly, 1 μl Lipofectamine 2000 (Thermo Fisher Scientific, 11668019) and 1 μg overexpression plasmid were mixed into 250 μl of Opti-MEM I Reduced Serum Medium (Merck, 31985062), respectively. Then the mixture was added to the cultured MuSCs for transfection and subsequent experiments were conducted at 24 or 48 h after transfection.
Western blot
Protein samples were extracted using western and IP (Immunoprecipitation) lysis buffer (Beyotime, P0013). The protein extracts were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), transferred to nitrocellulose membranes, and blocked with 5% BSA in TBST for 1 h. The membranes were then incubated overnight at 4 °C with anti-Mettl3 (Abcam, ab195352, 1:1000), anti-MyoD (Santa Cruz Biotechnology, sc377460, 1:200), or anti-GAPDH (Cell Signaling Technology, 2118S, 1:5000). Afterward, the membranes were incubated with the corresponding secondary antibodies at room temperature for 1 h. Signal detection was performed using GelDoc XR (Bio-Rad) and LumiQ ECL liquid (ShareBio, SBWB012).
m6A dot blot assay
RNA was heated at 95 °C for 3 min to disrupt the secondary structure, followed by cooling on ice for 3 min. A source imprint was left on the nitrocellulose membranes with the rough end of a 1 mL pipette tip to facilitate spiking. Two gradients of RNA (400 ng and 800 ng) were cascaded onto the membrane, air-dried for 5 min, and then UV cross-linked for 1 h. After blocking with 5% BSA in TBS for 1 h, the membranes were incubated with anti-m6A antibody (Abcam, 151,230) overnight at 4 °C. Following three washes with TBST and 1 h of incubation with secondary antibody at room temperature, chemiluminescence was utilized for signal detection.
Methylated RNA immunoprecipitation (MeRIP)–qPCR
MeRIP was performed with m6A RNA Methylation Fragment Enrichment kit (Epigentek, P-9018) following the manufacturer’s protocol. Initially, 20 μg of total RNA was subjected to m6A immunoprecipitation, with 1/10 of the sample reserved as the input control. To facilitate m6A RNA immunocapture, an immunocapture buffer consisting of RNA samples, m6A antibody, and affinity beads was vortexed at room temperature for 90 min. Subsequently, RNA fragmentation was achieved using a cleavage enzyme mix, followed by the addition of proteinase K and RNA purification solution to remove excess proteins and isolate m6A-containing RNA from the immunoprecipitated complex. Finally, the immunoprecipitated m6A RNA was recovered using elution buffer. The level of m6A methylation in MyoD was assessed by RT–qPCR with primers listed in Additional file 1: Table S2.
mRNA and protein stability assay
For mRNA stability assay, actinomycin D (Act-D, MCE, HY-17559, 5 μg/mL) was used to globally inhibit mRNA transcription, and then MuSCs were harvested at time points of 0, 3, 6, 9, and 12 h. RNA extraction was then conducted for RT–qPCR analysis to evaluate RNA degradation, with GAPDH employed as the normalization reference.
To evaluate protein stability, MuSCs were treated with 100 μg/mL cycloheximide (CHX, MCE, HY-12302) and harvested at 0, 3, 6, 9, and 12 h. Subsequently, MyoD and GAPDH expression was then assessed by western blot analysis.
Luciferase reporter assays
To validate the functional m6A methylation site in MyoD, either the wild-type or mutant CDS (Coding Sequence)of MyoD was inserted behind the F-luc coding region of the pmiRGLO vector (Miaoling Biology, P0198). Mettl3f/f MuSCs and Mettl3 KO MuSCs were transfected with pmiRGLO, pmiRGLO-MyoD-CDS-WT, pmiRGLO-MyoD-CDS-Mut1 (A to G mutation at position 228), or pmiRGLO-MyoD-CDS-Mut2 (A to G mutation at position 234) for 24 h. Subsequently, luciferase activity was quantified using the Dual Luciferase Reporter Assay kit (Yeasen Biotechnology, 11402ES60). Renilla luciferase (R-luc) was used to normalize the firefly luciferase (F-luc) activity.
Bulk RNA sequencing and analysis
The NEBNext Ultra RNA Library Prep kit from Illumina (New England Biolabs) was utilized to create RNA sequencing libraries. Subsequently, paired-end sequencing was executed on the NovaSeq X Plus sequencer with a 2× 150 bp read length. To ensure data quality, quality control procedures were undertaken on the raw paired-end reads through the applications of SeqPrep and Sickle. Differential expression analysis was conducted using DEGseq, with genes identified as significantly differentially expressed if the fold change was greater than 1.5 and the adjusted p-value was < 0.001 [29]. Furthermore, for deeper insights, Gene Ontology (GO) analysis was conducted using Goatools [30].
m6A sequencing (m6A-seq) data analysis
MeRIP sequencing raw data were obtained from the Gene Expression Omnibus (GEO) repository (accession no: GSE169432). Raw sequencing reads were first mapped to the reference mouse genome (mm10) using Hisat2 software. The mapped reads from the IP and input libraries were then analyzed using the R package exomePeak to identify significant m6A peaks and differential peaks, with a significance threshold of FDR (False Discovery Rate) ≤ 0.05. The IGV software was used for visualization.
In vivo muscle force analysis
In vivo muscle force analysis of tibialis anterior (TA) muscle was conducted using a 3-in-1 whole-animal system (Aurora Scientific; 1300A) [25]. After anesthetizing the mouse, the hind limbs were shaved and securely placed in a frame to ensure stability without hindering blood flow. Bread silk suture was used to ligate the distal TA muscle around the patellar ligament. An incision was made to expose the sciatic nerve, which was then tied off at its proximal end. The distal TA tendon suture loop was fastened to the lever arm hook of the instrument for measuring twitch and tetanus force. Each treatment was repeated for three times, and data analysis was performed using DMA software (Aurora Scientific).
Statistical analysis
The data were presented as mean ± SD from at least three independent experiments. Group comparisons were conducted employing either a two-tailed Student’s t-test or one-way ANOVA, with statistical analyses performed using GraphPad Prism 9 or SPSS version 26.0. A p-value < 0.05 was considered significant, with *p < 0.05, **p < 0.01, ***p < 0.001, and ns representing no significance.




留言 (0)