Animals and experimental design
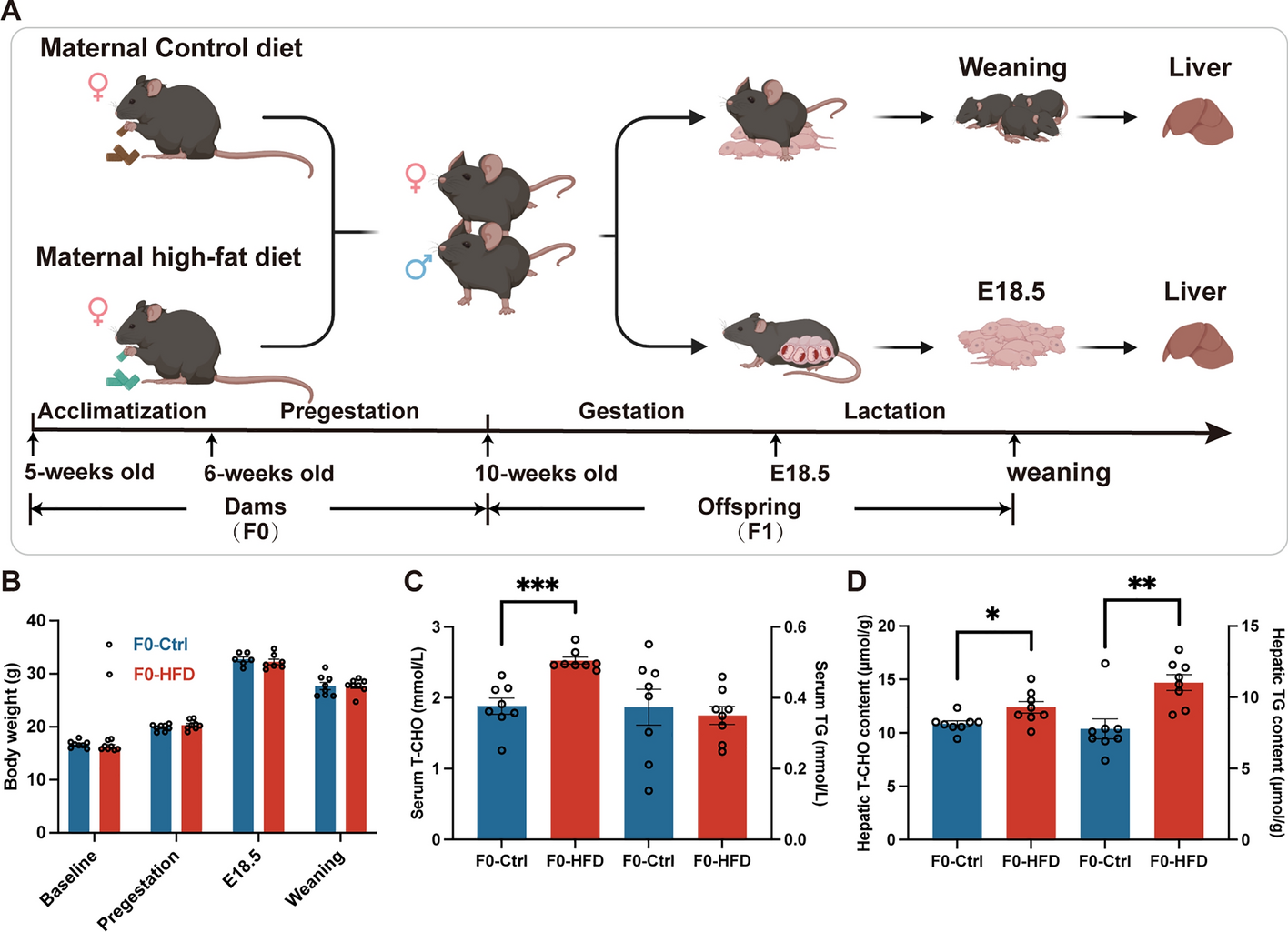
Five-week-old female C57BL/6 J mice were obtained and maintained under standard conditions at 22 ± 2 °C with a 12-h light/12-h dark cycle. Following 1-week acclimatization, 6-week-old female mice were randomly fed with a standard diet (13% fat, 24% protein, 63% carbohydrate, 3.44 kcal/g) (F0-Ctrl for dams) or a HFD (60% fat, 20% protein, 20% carbohydrate, 5.24 kcal/g) (F0-HFD for dams) for a duration of 4 weeks. Subsequently, 10-week-old female mice were mated with standard-diet-fed male mice (female:male = 2:1) to control for potential differences in sires. Observation of vaginal plugs was designated embryonic day 0.5 (E0.5). Throughout gestation and until pup weaning, females were maintained on their respective diets. Two cohorts of dams were established for this study. In the first cohort, dams were anesthetized on gestational day 18.5, and fetal livers were collected. Experiments were conducted on the fetus irrespective of the sex of the pups. In the second cohort, each dam had a litter size of 6–10, and the litter size was standardized to six mice per litter post birth to avoid nutritional bias, irrespective of the sex of the pups. At weaning, we randomly selected one male offspring from each litter for further analysis (F1-HFD and F1-Ctrl for offspring, respectively). A 12-h fast was followed by anesthesia and sacrifice, and blood and livers were immediately harvested and stored at −80 °C. The weights of subcutaneous and visceral adipose tissue were recorded.
Measurement of lipids concentrations
Serum total cholesterol (T-CHO), low-density lipoprotein cholesterol (LDL-C), and triglyceride (TG) were quantified using commercial kits (A111-1, A113-1, A110-1, respectively, Jiancheng Bioengineering Institute, Nanjing, China). Hepatic T-CHO and TG contents were extracted from liver samples and quantitated using the same commercial kits (A111-1, A110-1, respectively; Jiancheng Bioengineering Institute, Nanjing, China). Each sample was assayed in duplicate.
Oil Red O staining
Liver samples were embedded in Tissue-Tek O.C.T. compound and sectioned at 10 µm thickness for each mouse. Oil Red O staining was performed by immersing slides in 60% isopropanol for 2 min, followed by staining with working Oil Red O for 10 min (G1015, ServiceBio, Beijing, China). After brief rinsing with distilled water and 60% isopropanol, slides were stained with hematoxylin (G1004, ServiceBio, Beijing, China) for 15 s. Images were captured using an Olympus DP71 microscope.
Hematoxylin–eosin (H&E) staining and immunohistochemistry
For H&E staining, liver sections (5 μm) were sequentially incubated in hematoxylin for 15 s and eosin for 30 s (G1002, ServiceBio, Beijing, China). For immunohistochemistry, liver sections were pretreated in citrate buffer (PH 6.0, 98 °C) for 20 min, followed by quenching of endogenous peroxidase activity with 0.3% H2O2 for 15 min. After blocking in goat serum (C0265, Beyotime, Beijing, China) for 20 min, sections were incubated with primary antibodies against myocyte enhancer factor 2A (MEF2A) (1:1000, ab264329, Abcam, Cambridge, UK) and CYP7A1 (1:1000, 18054–1-AP, Proteintech, Wuhan, China) overnight at 4 °C. The next day, sections were incubated with a secondary antibody (PV9000, Zhongshan Gold Bridge Biotechnology Co, Beijing, China) for 1 h at room temperature. Visualization was achieved using diaminobenzidine (DAB) staining (ZLI9018, Zhongshan Gold Bridge Biotechnology Co, Beijing, China), followed by counterstaining with hematoxylin. Images were captured with an Olympus DP71 microscope. Five random fields were selected from each section, and mean integrated optical density (IOD/area) was quantified using Image-Pro Plus 6.0 software.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from liver tissues or cells using TRIzol reagent (15596026, Invitrogen, Waltham, MA, USA), and 1 µg of RNA was reverse transcribed into cDNA using the high-capacity cDNA reverse transcription kit (4375222, Thermo Fisher Scientific, Hudson, NH, USA). Gene expression was analyzed using SYBR Green (A25742, Thermo Fisher Scientific, Hudson, NH, USA). β-Actin was used as an endogenous control. Primer sequences are listed in Table 1.
Table 1 Primer sequences of genes for quantitative RT-PCR analysisWestern blots
Hepatic tissues and cells were lysed in RIPA lysis buffer (PLB004N-BR100, Snodetech, Beijing, China). Total protein was separated by 10% SDS-PAGE gel and transferred onto nitrocellulose membranes. After blocking for 1 h in 5% fat-free milk, membranes were incubated overnight at 4 °C with primary antibodies against DNA methyltransferases 1 (DNMT1) (1:1000, #5032, Cell Signaling Technology, Danvers, MA, USA), DNMT3A (1:1000, 20,954–1-AP, Proteintech, Wuhan, China), DNMT3B (1:1000, 26,971–1-AP, Proteintech, Wuhan, China), MEF2A (1:1000, Ab76063, Abcam, Cambridge, UK), CYP7A1 (1:1000, 18,054–1-AP, Proteintech, Wuhan, China), and β-actin (1:1000, #4970 s, Cell Signaling Technology, Danvers, MA, USA). After washing, membranes were incubated with a secondary antibody (1:5000, Zhongshan Gold Bridge Biotechnology Co, Beijing, China) for 1 h at room temperature. Protein bands were visualized using an enhanced chemiluminescence (ECL) detection kit. β-Actin was used as an endogenous control.
RNA sequencing (RNA-seq) analyses
RNA was quantified using the ND-2000 (NanoDrop Technologies). To construct sequencing libraries, the OD260/280 ratio (1.8 × 2.2), the OD260/230 ratio (2.5 × 2.0), the RIN, and the 28S:18S ratio (1.0 × 1.0) of the RNA samples (> 1ug) were tested. Following the manufacturer’s instructions (Illumina, San Diego, CA), RNA was purified, reverse transcribed, library constructed, and sequenced at Shanghai Majorbio Biopharm Biotechnology Co., Ltd.. A HISAT2 alignment was performed on clean reads to determine their alignment to the reference genome [23]. Each sample’s mapped reads were assembled using StringTie using reference data [24]. Differential expression analysis was performed using the DESeq2, which is a software package for estimating variance−mean dependence and testing for differential expression based on a negative binomial model [25]. Differentially expressed genes (DEGs) with |log2FC|> 0 and P < 0.05 were considered to be significant, as in previous studies [26, 27]. Functional-enrichment analyses including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) were performed to identify which DEGs were significantly enriched in GO terms and metabolic pathways, at Bonferroni-corrected P value < 0.05, compared with the whole-transcriptome background. GO functional enrichment and KEGG pathway analysis were carried out by Goatools and Python SciPy software, respectively. Predicting possible upstream transcription factors of DEGs was performed using bioinformatics databases, including Animal Transcription Factor Database (AnimalTFDB, http://bioinfo.life.hust.edu.cn/AnimalTFDB4/) and ChIP-X Enrichment Analysis 3 (ChEA3, https://maayanlab.cloud/chea3/).
Methylation analysis
Hepatic genomic DNA was extracted using the E.Z.N.A. DNA kit (Omega Bio-tek, Inc., Norcross, GA). Promoter methylation of the target gene was detected using the MassARRAY EpiTYPER platform (Agena Bioscience, San Diego, CA, USA). The target region of Mef2a was amplified with specific primers. The forward sequence of the primer was 5′-aggaagagagGTTGTGGTATAAGGATAAGAGGGG-3′ and reverse sequence of the primer was 3′-cagtaatacgactcactatagggagaaggctTAACCCAACAACTATCCAAACCTAA-5′. DNA methylation analysis was performed using MassARRAY EpiTYPER software (Agena Bioscience, San Diego, CA, USA).
Cell culture
Human hepatocellular carcinoma cell lines (HepG2) (SCSP-510) and H9C2 cells (SCSP-5211) were obtained from the Chinese National Infrastructure of Cell Line Resource. HepG2 cells were cultured in MEM medium (11090081, Invitrogen, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS), 1% nonessential amino acids (11140050, Invitrogen, Waltham, MA, USA), 1% glutamax (35050061, Invitrogen, Waltham, MA, USA), 1% sodium pyruvate 100 mM solution (11360070, Invitrogen, Waltham, MA, USA), and 1% penicillin–streptomycin (15140122, Gibco, Life Science, Pittsburgh, PA, USA), and maintained at 37 °C with 5% CO2. H9C2 cells were cultured in DMEM medium (10566016, Gibco, Life Science, Pittsburgh, PA, USA) supplemented with 10% FBS and 1% penicillin–streptomycin. For LDL-C treatment, HepG2 cells were incubated with 200 mg/dl LDL-C for 24 h (YB-001, Yiyuan biotech, Guangzhou, China). For 5-azacitidine (5-AZA) treatment, HepG2 cells were incubated with 0 mmol/L to 1 mmol/L 5-AZA for 24–48 h (A2385, Sigma, Steinheim, Germany).
Transient knockdown experiment
Human MEF2A siRNA and scrambled siRNA were designed and synthesized by GenePharma (Shanghai, China). The sequences of siRNA targeting Mef2a were used as GGGCAGUUAUCUCAGGGUUTT (sense sequence) and AACCCUGAGAUAACUGCCCTT (antisense sequence), and the sequences of scramble siRNA were UUCUCCGAACGUGUCACGUdTdT (sense sequence), and ACGUGACACGUUCGGAGAAdTdT (antisense sequence). Quantities of 50 nM of each siRNA duplexes were transfected into cells growing in 12-well plates using Lipofectamine RNAiMAX (13,778–030, Invitrogen) for 24 h according to the manufacturer’s instructions. The silencing efficiency was verified by qRT-PCR and western blot, with scrambled siRNA used as a negative control.
Generation of stable MEF2A overexpression HepG2 cell line by lentivirus
A stable HepG2 cell line overexpressing MEF2A was established using a lentiviral-based delivery system. MEF2A and control plasmids were synthesized by Ubigene (Guangzhou, China). HEK293T cells were seeded in a 10-cm culture dish. Lentiviral and packaging vectors (pCMV-VSVG:psPAX2:target plasmid = 5 μg:15 μg:10 μg) were transfected into the cells on the following day. After 48 h, the cells were harvested and filtered through a 0.45-µm membrane. Lentiviral particles were supplemented with 5 μg/mL polybrene. Afterward, lentiviral particles were added to HepG2 cells and incubated for 24 h. The cells were then supplemented with fresh medium and further incubated for 48 h. Stable cells were selected using puromycin (1 µg/mL) for the following experiments.
Lucia luciferase activity assay for DNA methylation
To investigate the regulatory effect of CpG methylation in the Mef2a promoter region in vitro, a human promoter containing 2000 bp of Mef2a was inserted into a reporter plasmid CpG-free basic Lucia (pcpgf-baslc, InvivoGen, San Diego, CA, USA), which lacks CpG sites. The recombinant Mef2a plasmid was methylated using M.SssI (M0226S, NEB) according to the manufacturer’s instructions, and then purified and recovered using the Monarch PCR and DNA Cleanup Kit (T1030S, NEB). H9C2 cells were transfected with 200 ng of unmethylated or methylated pCpG free-basic-Lucia plasmid along with 10 ng of firefly luciferase reporter vector. Lucia luciferase activity was measured with QUANTI-Luc (rep-qlc4lg1, InvivoGen, San Diego, CA, USA). The firefly luciferase activity was measured using the Bio-Lite Luciferase Assay System (DD1201-01, Vazyme, Nanjing, China). Lucia luciferase activity was normalized against firefly luciferase activity.
Dual luciferase reporter assay
The Cyp7a1 promoter-luciferase reporter construct containing 2.0 kb of the H_Cyp7a1 promoter sequence (-1950 to +50) and the pRL Renilla luciferase control reporter vector was constructed by Genomeditech (Guangzhou, China). HepG2 cells were seeded in 12-well plates, and then transfected with 200 ng of Cyp7a1-luciferase reporter and 20 ng pRL Renilla along with 200 ng of Mef2a plasmid. Luciferase activity was determined 48 h post-transfection using the Dual-Luciferase Reporter Assay System (E1910, Promega Corp, WI, USA). Relative luciferase activity was normalized against Renilla luciferase activity.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 10.0 software. Data are presented as mean ± standard error (SEM). Student’s unpaired t-test was used for comparisons between the two groups, while one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was utilized for multiple comparison analysis. P value < 0.05 was considered statistically significant.




留言 (0)