Animals and treatments
All animal studies were conducted under ethical policies and approved by the Animal Care and Use Committee at the Institute of Laboratory, Guangzhou Medical University (approval no. G2023-147, 2/3/2023). Female Institute of Cancer Research (ICR) mice (6–8 weeks old) and NOD/LtJ mice strain (N000235) (6–8 weeks old) were obtained from Gempharmatech (Nanjing, China). All mice were housed in a pathogen-free animal facility maintained under standard conditions and acclimatized to their environment for 7 days before starting experiments.
The mice were randomly divided into four groups, each containing six mice, including (I) control group, ICR mice given a basic diet and vehicle (0.5% sodium carboxymethyl cellulose (CMC-Na)); (II) model group, NOD/LtJ mice given a basic diet and vehicle; (III) genistein (50 mg/kg) group, NOD/LtJ mice given genistein (50 mg/kg) prepared with 0.5% CMC-Na solvent; (IV) hydroxychloroquine (HCQ, 50 mg/kg) group, NOD/LtJ mice given HCQ (50 mg/kg) prepared with 0.5% CMC-Na solvent. Respective formulations were administrated by gavage in all four murine groups for 8 weeks. Finally, followed by sacrifice, the peripheral blood from the ophthalmic vein was collected, and submandibular gland tissues were rapidly collected for analysis.
Water intake and saliva flow rate assessment
Water intake was recorded weekly after drug administration; it was measured as water intake (mL) divided by body weight (g), while saliva flow rate was measured biweekly after an overnight fast as described previously [27]. Briefly, mouse was anesthetized with pilocarpine hydrochloride (0.1 mg/kg i.p.), then weight increase (mg) was calculated divided by body weight (g) during 10 min.
Hematoxylin–eosin (H&E) staining and immunohistochemistry
The submandibular gland tissues embedded in paraffin were cut into 4-μm-thick slices and then stained with hematoxylin and eosin (H&E) to evaluate inflammatory cell infiltration and histopathological damage. For immunohistochemistry (IHC), the tissue sections were first blocked for 30 min, then incubated with primary antibodies, including anti-aquaporin 5 (AQP5, Abcam, ab305304) at a dilution of 1:100, and anti-ACSL4 (Santa Cruz, SC-365230) at a dilution of 1:50 at 4 ℃ for 16 h. The following day, slices were washed with PBS, followed by addition of the secondary antibody for 1 h at room temperature, followed by diaminobenzidine (DAB) solution incubation for 1 min and counterstaining with hematoxylin. Quantification of representative images at 100× magnification was carried out by an independent observer, Additionally, automated quantification was conducted using Image-Pro Plus software (version 6.0).
Enzyme-linked immunosorbent assay
The serum levels of IFNγ, anti-SSA/Ro, and anti-SSB/La autoantibodies were detected using enzyme-linked immunosorbent assay kits (Mlbio, Shanghai, China) following the manufacturer’s instructions.
Real-time quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using Trizol reagent, and complementary DNA (cDNA) was synthesized using the Evo M-MLV reverse-transcribed kit (Accurate Biology, Hunan, China) following the manufacturer’s protocol. The cDNA was subjected to RT-qPCR analysis using SYBR Green Pro Taq HS mix (Accurate Biology, Hunan, China). β-Actin was used as a standard control to analyze the relative expression mRNA levels according to the 2(−ΔΔCt) method. All primer sequences are listed in Supplementary Table S1.
RNA sequencing data analysis
RNA sequencing was performed at Biowefind (Wuhan, China). Raw data underwent processing with the robust multiarray mean algorithm (RMA) within the “Affy” package. Differentially expressed genes (DEGs) were identified using linear models from the “LIMMA” package in R language. Gene function classification and evaluation of biological functions were carried out using the Bioinformatics online tool. Heatmap, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment, and Gene Ontology (GO) were performed to analyze the role of DEGs. A significance level of p < 0.05 was applied. Receiver operating characteristic (ROC) analysis was conducted using the “pROC” package in R to predict the diagnostic validity of biomarkers.
RNA fluorescence in situ hybridization (FISH) assay
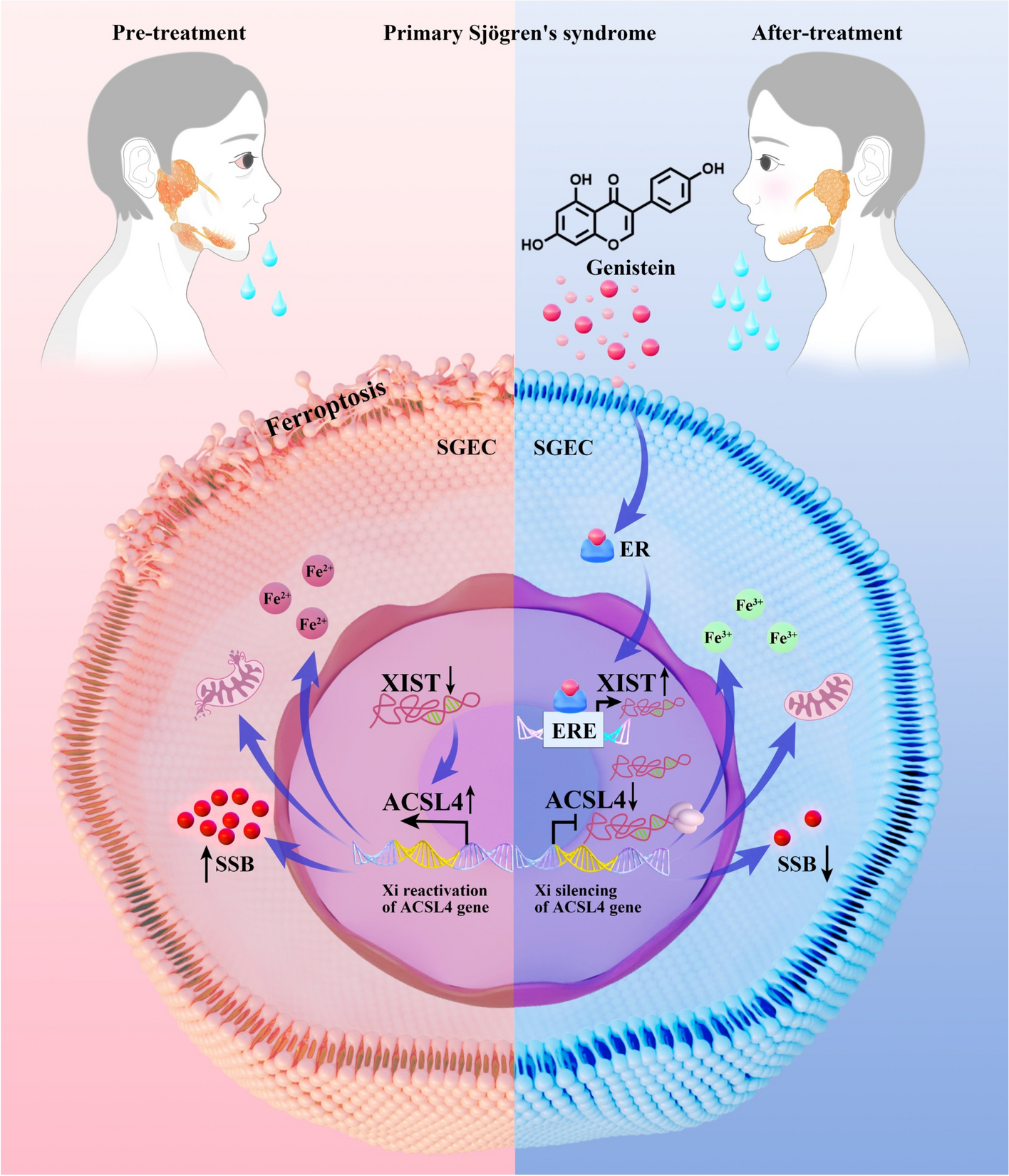
For RNA FISH, we used the Xist/XIST and Acsl4/ACSL4 probes designed by GenePharma Co., Ltd. (Shanghai, China), with sequences listed in Supplementary Table S2. Xist and Acsl4 RNA FISH in submandibular gland tissue or XIST and ACSL4 RNA FISH in SGECs was performed using RNA FISH SA-Biotin kits (GenePharma) according to the manufacturer’s recommendations. Finally, slides were visualized using a fluorescence microscope (Olympus BX43, Olympus, Tokyo, Japan).
Flow cytometry
Cells were incubated with fluorescent-conjugated antibodies for 15 min on ice to stain surface markers, then washed with cell staining buffer (BioLegend, San Diego, USA). Subsequently, Cells were fixed and permeabilized using a transcription-factor staining buffer, followed by a 30 min of incubation at room temperature with fluorescent-conjugated antibodies to stain intracellular antigens. The antibodies used for flow cytometry were APC anti-mouse FOXP3 (#32,007), FITC anti-mouse CD4 antibody (#100,411), and PE anti-mouse IL-17A (#116,107). All antibodies were purchased from BioLegend.
Surface plasmon resonance analysis (SPR)
A Biacore 8K system (Cytiva, Marlborough, MA, USA) was used to analyze the direct interaction between ERα and genistein. ERα recombinant protein was immobilized on Series S Sensor Chip CM 5 (GE Healthcare Life, Chicago, USA) according to the manufacturer’s instruction. After that, different concentrations of genistein (12.5–200 μmol/L) were diluted in a running buffer and injected into the system as the analyte. The parameters for SPR were as follows: flow rate, 30 µL/min; association time, 60 s; dissociation time 90 s; temperature, 25 °C. Finally, the interaction parameters were obtained using Biacore evaluation software (version 2.0).
Molecular docking and molecular dynamics (MD) simulations
We used AutoDock Vina for molecular docking. Initially, the ERα was prepared by deleting water molecules and bound ligands using the PyMol software. Subsequently, hydrogens were added, and energy minimization was performed using AutoDock. Finally, flexible ligands docking into the rigid binding site was conducted within the “Grid” module, and the Grid scores were used to estimate and rank ligand binding energies [28]. The optimal conformation with the lowest binding energy was selected as the initial conformation for MD simulations using Gromacs 2022.3 software as before [29]. The root-mean-square variance (RMSD), root-mean-square fluctuation, and protein rotation radius of each amino acid trajectory were calculated.
Cell culture
The human SGEC-line A253 (ATCC Number: HTB-41) was purchased from Zhejiang Meisen Cell Biotechnology Co., Ltd. (ATCC no. HTB-41) (Zhejiang, China). Cells were cultured in RPMI-1640 (Hyclone, USA) medium supplemented with 10% fetal bovine serum (FBS, Gibco, USA) in a humidified atmosphere with 5% CO2 at 37 °C. The cells were passaged every three days.
Cell viability assay
The assessment of cell viability in SGECs was conducted using Cell Counting Kit-8 (CCK-8) assays following the manufacturer’s protocols (MCE, Shanghai, China). These were performed at different concentrations of genistein (ranging from 0.001 µM to 100 µM), at specific time points of 24 h, 48 h, and 72 h post genistein intervention.
Chromatin immunoprecipitation (ChIP) assay
The SGECs ChIP assay was performed according to the manufacturer’s protocol (BersinBio, Guangzhou, China). Sonicated lysates were incubated with 4 µg ERα (ab32063, Abcam) and mixed overnight at 4 °C. The next day, antibody-bound chromatin was incubated with Protein-G, washed with radioimmunoprecipitation assay (RIPA) buffer, and eluted in the elution buffer. The eluted sample was incubated at 65 °C overnight to reverse the crosslinking, and DNA was extracted using a TIANquick Midi purification kit (TIANGEN, DP204, Beijing, China) and subjected to PCR analysis. The primers ERE1 and ERE2 (Supplementary Table S3) were designed to amplify the XIST promoter region that contains ERα binding sites from the Jaspar database. After amplification, PCR products were resolved on a 1.5% agarose gel.
Western blotting
Cells were lysed using RIPA buffer (Thermo Fisher Scientific, Waltham, MA, USA) containing protease inhibitors and phosphatase inhibitors (Beyotime, Nanjing, China), and protein concentrations were measured using a BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Total protein extracts (30 µg) were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) (Epizyme, Shanghai, China) using polyacrylamide gels and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Burlington, MA, USA). After being blocked with 5% skim milk for 1 h at room temperature, PVDF membranes were incubated with primary antibody overnight at 4℃. Then, they were incubated with secondary antibody for 1 h at room temperature. Finally, the protein was visualized by enhanced chemiluminescence reagents (Elabscience Biotechnology, Wuhan, China). Antibodies used in western blotting included: β-actin (ACTB, 1:10,000, Proteintech, 66,009–1-Ig), AQP5 (1:500, Santa Cruz, sc-514022), SSB (1:1000, Proteintech, 11,720–1-AP), and HRP-goat anti-rabbit/mouse secondary antibody (1:5000, Proteintech, RGAR001, RGAM001).
Short interfering RNA (siRNA), short hairpin RNA (shRNA), and overexpression RNA (OE RNA) transfection
SiRNA, shRNA, OE RNA, and negative controls were designed and synthesized by GenePharma Co., Ltd. (Shanghai, China). To study the effect of XIST knockdown on ACSL4 and SS-like symptoms, 20 nM of siRNA targeting XIST was transfected into SGECs using EZ Trans (Life-ilab, Shanghai, China). For validation of ACSL4 inactivation by XIST-mediated gene silencing, 20 µg sh-split ends (SPEN) plasmid was transfected into SGECs. To validate the effect of ACSL4 in SS, sh-ACSL4 or OE-ACSL4 plasmid was transfected into SGECs. The method for using siRNA, sh-RNA, or OE-RNA transfection was done as specified by the manufacturer. Transfected cells were used for the experiments. The sequences of siRNA and plasmids used here are listed in Supplementary Table S4.
Measurement of malondialdehyde (MDA) and glutathione (GSH)
At the end of the experiments, SGEC lysates were collected. In these lysates, MDA and GSH were assessed by using the MDA (S0131, Beyotime) and GSH detection kits (S0103, Beyotime) according to the manufacturer’s instructions.
Transmission electron microscopy (TEM) assay
SG and SGEC samples were fixed in 2.5% glutaraldehyde and sent to Servicebio (Wuhan, China) for sample preparation and image acquisition.
Detection of intracellular Fe2+ levels in SGECs
Intracellular Fe2+ levels were detected using the FerroOrange probe (Dojindo, Tokyo, Japan) following the manufacturer’s instructions.
Statistical analysis
All experiments were repeated at least three times. Data are shown as mean ± standard deviation (mean ± SD). Statistical analysis was performed using t-tests for comparison of two groups, and using one-way analysis of variance (ANOVA) followed by post hoc comparison for comparison between multiple groups. p < 0.05 was considered statistically significant. GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA) was used for statistical analysis and figure preparation.




留言 (0)