
記住我

Male Sprague-Dawley (SD) rats (230–250 g, 6 ~ 8 weeks old) were purchased from Hunan SJA Laboratory Animal Co., Ltd (Changsha, PR China). The SD rats were raised in the specific pathogen-free (SPF) unit (three per cage) of the experimental animal center of South China Agricultural University, Guangzhou, China [license number: SYXK (Guangdong) 2022 − 0136]. All rats were housed under a 12-h:12-h light/dark cycle and 40-70% relative humidity in a temperature-controlled room (20–26 °C) with free access to food and water. All surgical procedures and animal care were conducted following the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals, also approved and reviewed by the South China Agricultural University Animal Ethics Committee (Approval no. 2022D148). A total of 250 rats were randomly assigned to five groups (n = 50 per group).
Cell cultureHuman umbilical cord MSCs were provided by the Biotherapy Centre of the Third Affiliated Hospital of Sun Yat-sen University. Specifically, MSCs were successfully isolated and cultured in vitro from the umbilical cords of six healthy donors, who provided informed consent. The MSCs were cultured in a serum-free medium (Yocon, China) at 37 °C in a humidified incubator with 5% CO2 and 95% air. The medium was changed every 2 days commonly. When reaching 90% confluence, cells were detached by incubating with accutase (Thermo Fisher, A1110501, USA) for 5 min at 37 °C and then replated for continuous passage. MSCs within 6 cell passages were used for all experiments.
PC12 cell line (iCell) purchased from Guangzhou Taylor Biotechnology Co., LTD. Cells were cultured in 90% 1640 medium(GIBCO, USA) supplemented with 10% fetal bovine serum (FBS) (Procell, China), 100 IU/mL penicillin, and 100 µg/mL streptomycin (Thermo Fisher, USA) at 37 °C in a humidified incubator with 5% CO2 and 95% air. The medium was changed every 2 days. Following washed by phosphate-buffered saline (PBS)(GIBCO, USA) for 1 min, cells were detached by incubating with 0.25% trypsin (supplemented with EDTA) for 1 ~ 2 min at 37 °C when reaching 90% confluence and then replated for continuous passage.
Induction and treatment of tMCAOThe ischemic stroke model was induced by the right side of transient middle cerebral artery occlusion (tMCAO) as previously described [23, 24]. The tMCAO model is widely utilized in ischemic stroke research for high reproducibility and without craniotomy [25, 26]. Briefly, rats were anesthetized using 2% pentobarbital sodium (50 mg/kg, Sigma-Aldrich) via intraperitoneal injection. Afterward, the right common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA) were exposed during the surgery. A filament (CINONTECH, China, A4-263650) was inserted into the ICA to block blood flow from the origin of the middle cerebral artery. The duration of cerebral ischemia was 60 min. We have made all efforts to minimize animal suffering, including oral ibuprofen (60 mg/kg) (Macklin, China, I821809-25 g) to manage post-surgery pain and 1 h of constant temperature management (37 °C) using a heating pad (20 × 35 cm; G-CLONE, China, VG-JRD-S) during anesthesia [27, 28]. After evaluation with modified neurological severity score (mNSS) [29], rats in the sham group with scores of 0 and tMCAO rats with moderate impairment (7 ~ 12 scores) were selected for this study, indicating the success of tMCAO establishment. Rats with mild impairment (1 ~ 6 scores) or severe impairment (13 ~ 18 scores) were excluded from the study. The mortality rate of tMCAO was less than 5% 24 h post-surgery, according to our experience, which is consistent with previous studies [26, 30].
For the treatment regimen, experimental rats were randomly assigned to 5 groups: Control group (tMCAO): 500 µL PBS(GIBCO, USA) was injected into the caudal vein; Repeated transcranial magnetic stimulation group (rTMS): 500 µL PBS was injected via a caudal vein and intervened with rTMS 1 d after tMCAO one time per day for 14 consecutive days; MSCs transplantation group (MSCs): 2 × 10^6 MSCs (per rat) were injected; Combination group (MSCs + rTMS): rats were injected with 2 × 10^6 MSCs (per rat) and treated with rTMS 4 h following MSCs transplantation one time per day for consecutive 14 d; sham group (sham): no 4 − 0 nylon suture was inserted, and the same volume of PBS was injected into the caudal vein as in the tMCAO group.
Differentiation, identification, and administration of MSCsOsteogenic and lipogenic differentiation were conducted in vitro to assess the multidirectional differentiation potential. Briefly, human umbilical cord MSCs were seeded in a low glucose-DMEM (L-DMEM) (GIBCO, USA) complete medium prior to differentiation induction. Once the cells reached 80 ~ 90% confluence, the medium was replaced with bone induction medium containing L-DMEM, 10% FBS, 2 mM glutamine (GIBCO, USA), 100 IU/mL penicillin, 100 mg/mL streptomycin, 0.1 µM dexamethasone (Merck, USA), 50 µg/mL ascorbic acid (Sigma-Aldrich, USA) and 10 mM β-glycerol phosphate (Sigma-Aldrich, USA). After 2 ~ 3 weeks, osteogenic differentiation was confirmed by the mineralization of extracellular matrix and calcium deposits assessed using 0.5% Alizarin Red S (Sigma-Aldrich, USA) staining. Adipogenic differentiation was induced using adipogenic induction medium containing high glucose (H-DMEM) (GIBCO, USA), 10% FBS, 2 mM glutamine, 100 IU/mL penicillin, 100 mg/mL streptomycin, 1 µM dexamethasone, 10 µg/mL insulin (Prospect, Israel), 0.5 mM isobutylmethylxanthine (IBMX) (Sigma-Aldrich, USA) and 0.2 mM indomethacin (Sigma-Aldrich, USA) once the cells reached 100% confluence. Adipogenic differentiation was verified by the typical production of lipid droplet staining with Oil Red O (Sigma-Aldrich, USA).
The third passage human umbilical cord MSCs cultured in serum-free medium were digested with accutase at 37 ℃, resuspended to a concentration of 1 × 10^6/mL cell suspension, filtered through 70 μm sieve, and transferred to flow tubes, centrifuged for 5 min at 4 ℃, 450 × g, and resuspended in 100 µL PBS. Flow cytometric analysis was performed on LSR II (BD, USA) or CytoFLEX flow cytometer (Beckman Coulter, Fullerton, CA, USA), and data were analyzed using FlowJo7.6 software (Treestar, Ashland, USA). The utilized antibodies are listed in Supplementary Tables 1, and the corresponding isotype control antibodies were purchased from BD Bioscience. These antibodies were employed to label human umbilical cord MSCs at room temperature for the detection of cell phenotype via flow assay.
Human umbilical cord MSCs with sixth passage were digested with 0.25% trypsin (GIBCO, USA) at 37 ℃, and resuspended in PBS with a density of 4 × 10^6/mL MSCs. And 500 µL cell suspension (2 × 10^6/mL MSCs) was administrated per rat via a caudal vein 24 h after tMCAO surgery, according to previous studies [31, 32]. In brief, the rats were secured in a restrainer (Xiangbo, China, XB-DSL) measuring 230 mm (height) × 70 mm (outer diameter) × 60 mm (inner diameter), with the caudal vein exposed. The tail was wiped with alcohol for disinfection and to enhance the visibility of the caudal vein. An insulin syringe (BD, U-40, USA) was used to inject the MSC suspension into the caudal vein over a period of 10 to 15 s. After withdrawing the needle, pressure was applied to stop the bleeding, and the rats were released from the restrainer. Their vital signs were monitored for 30 min to ensure they were stable.
rTMS interventionIn vivo, a customized magnetic stimulator (CCY-IA, Wuhan Yiruide Medical Equipment, Wuhan, China) was used in this study to stimulate rats in the rTMS group and MSCs + rTMS group. The treatment was administered from 24 h to 14 days after tMCAO operation. All procedures followed a protocol described in previous studies [33]. Briefly, each rat was placed into a breathable rodent restraint bag (DecapiCones, Brain three Scientific, Braintree, MA, USA). A round prototype coil (6 cm in diameter with 3.5-T peak magnetic welds) was positioned perpendicular to the cortex on the surface of the cortical projection area of the ipsilateral primary motor cortex (right M1 zone) of each rat. The stimulation area of this coil covered a 1 ~ 2 cm2 area of the brain, covering the peri-infarct region [34]. The rTMS group was applied at 10 Hz, with 40 pulses per train, 10 s intertrain interval, and a total of 30 trains (1,200 pulses) for 7 min. The stimulation intensity was set to 26% of the maximum output strength of the machine, which is equivalent to 100% of the resting motor threshold (RMT). Motor-evoked potentials (MEPs) were measured at the right hind limbs and quadriceps femoris muscle using electromyography (MedelecSynergy; Oxford Instruments, Surrey, United Kingdom), as previously described [35]. The RMT was defined as the lowest stimulator output at which the peak-to-peak amplitude of the MEP was greater than 5% of its maximal amplitude in at least half of the 10 trials.
In vitro, followed by 24-hour reoxygenation, PC12 cells in 12-well plates were treated with rTMS every 8 h for 48 h. TMS treatment was conducted with a MagPro X100 magnetic stimulator (The MagVenture Company, Denmark) with a flat coin. The stimulation parameters were referred to a previous study [36]. Briefly, PC12 cells in the rTMS group and MSCs + rTMS group were applied at 10 Hz, 30% maximum output intensity of the machine, with 20 pulses per train, 10 s intertrain interval, and a total of 60 trains (1,200 pulses) for 11 min 44 s.
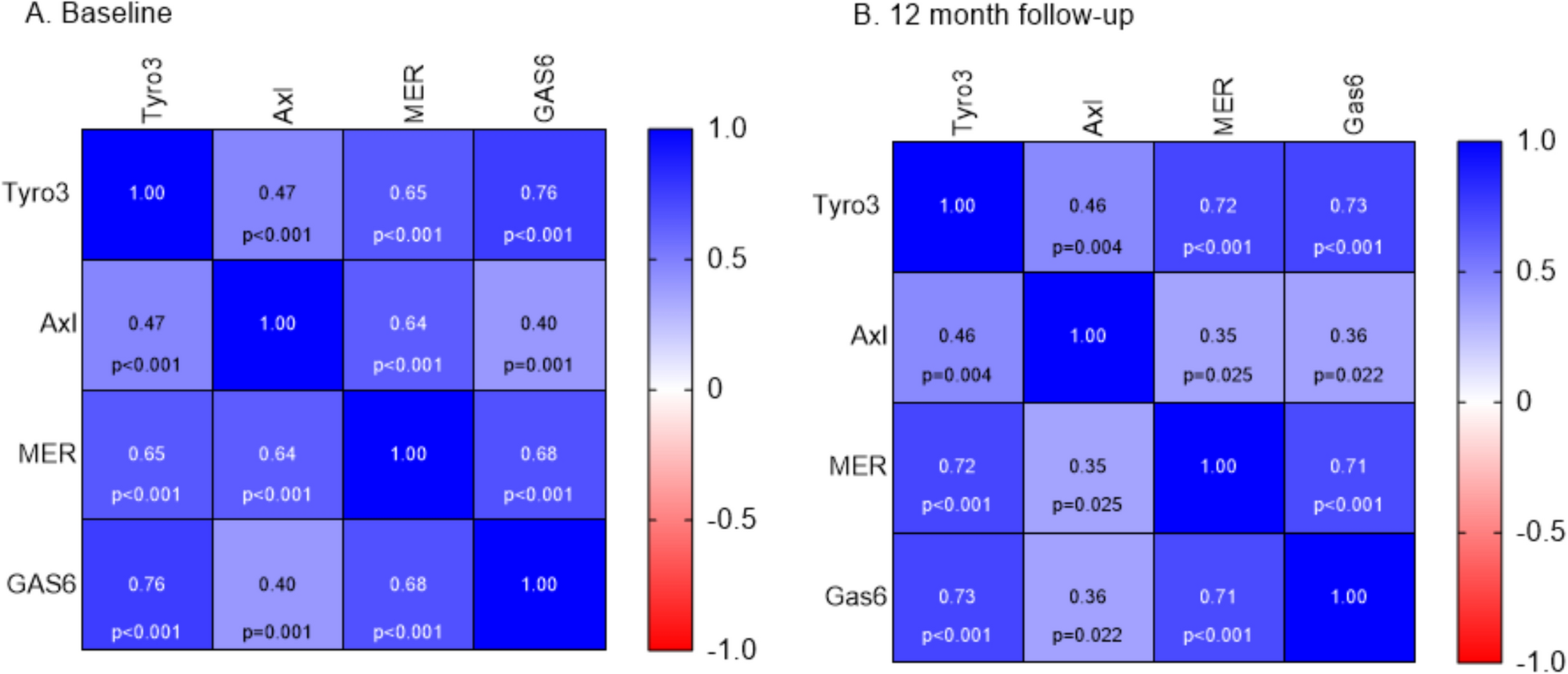
Fig. 1
Combining MSCs with rTMS treatment ameliorates neurological deficits in post-ischemic rats. (A)(a), Experimental design: MSCs transplantation was performed 24 h after the stroke. Behavioral tests were assessed at the indicated time points. rTMS was administrated following MSCs transplantation one time per day until animals were sacrificed. A(b), Schematic diagram of three behavioral tests. (B), The survival rates of each group were followed up every 24 hours for 14 days. n = 20 per group (C), The body weight of each group was tested at 0, 1, 4, 7, and 14 days post-stroke. n = 8 per group. (D), Neurological deficits were evaluated by the mNSS test for each group at 0, 1, 4, 7, and 14 days post-stroke. n = 8 per group. (E), Motor dysfunction was assessed by the cylinder test for each group at 0, 1, 4, 7, and 14 days post-stroke. n = 8 per group. (F), Sensorimotor dysfunction was assessed by the adhesive removal test up to 14 days post-surgery. Data were expressed as the latency to contact (F)(a) and remove (F)(b) tape from the contralateral forepaws. n = 8 per group. Asterisks, according to their color, indicate significant pairwise differences, *P < 0.05, ** P < 0.01 vs. tMCAO group; #P < 0.05 vs. rTMS group; &&P < 0 0.01 vs. MSCs group by two-way repeated-measures ANOVA and Tukey post hoc
Survival rate and behavioral testsThe survival rate was documented every 24 h from the tMCAO surgery day until 14 days. We used the Zea Longa test, the Bederson test, the mNSS test, the cylinder test, and the adhesive removal test to assess the neurological deficit of rats before tMCAO surgery (0 days), and at 1, 4, 7, and 14 days after tMCAO in a blinded fashion. The experimenter was blinded to the group allocation.
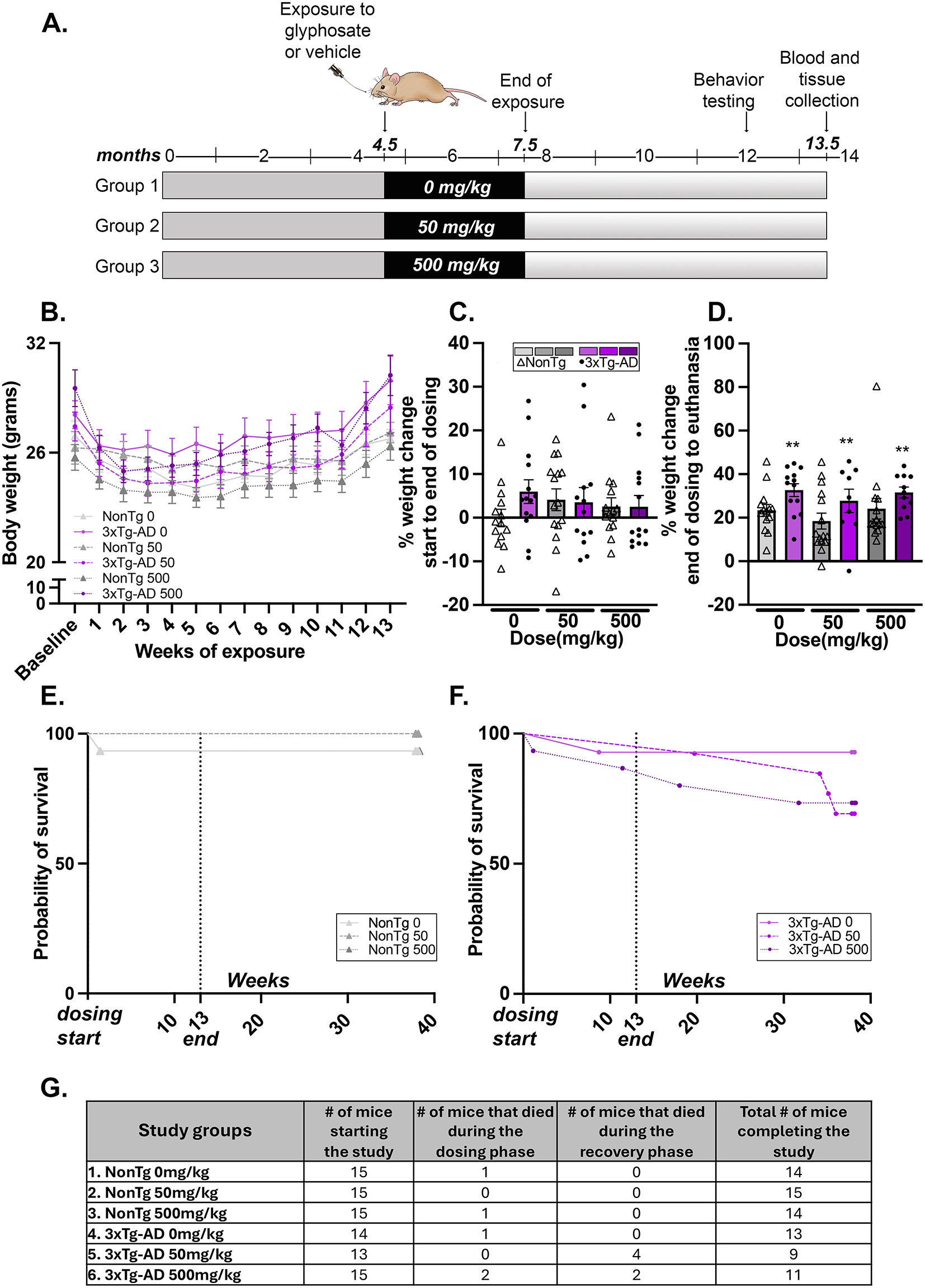
Fig. 2
MSCs combined with rTMS therapy reduce neuronal damage in the subacute phase after stroke. (A), Representative TTC staining images of the coronal brain sections of rats in the sham group, tMCAO group, rTMS group, MSCs group, and MSCs + rTMS group 4 days after tMCAO. (B), Bar graph showed quantification of infarct volume determined by TTC staining on day 1, day 4, and 7 after tMCAO surgery. n = 8 per group. (C), Diagram of the regions of the ischemic core and the peri-infarct region. (D), Neuronal degeneration in tMCAO rats with rTMS or/and MSCs therapy. Neurons (yellow arrows) and degenerating neurons (red arrows) were visualized using Nissl staining of brain sections in each group. The scale bar represents 50 μm. (E), Quantification of neurons was conducted in six randomized fields. n = 3 per group. (F), Quantification of degenerating neurons was conducted in three randomized fields. n = 3 per group. *P < 0.05, ** P < 0 0.01, *** P < 0 0.001 by one-way ANOVA and Tukey post hoc
The zea longa testThe Zea Longa test is a widely used method for assessing neurological deficits in animal models of stroke [26]. Briefly, neurological findings are evaluated on a five-point scale at 4 days post-stroke: a score of 0 indicates no deficits, while a score of 1 represents a mild focal deficit characterized by an inability to fully extend the left forepaw. A score of 2 indicates a moderate deficit, with the animal circling to the left, and a score of 3 reflects a severe deficit, where the rat falls to the left. Rats receiving a score of 4 do not walk spontaneously and exhibit a decreased level of consciousness.
The bederson testThe Bederson score test [37] involves lifting the rats’ tails to a height of 10 cm above the table surface at 4 days post-stroke. At this point, normal rats will have their forelimbs extended, while animals with neurological deficits may exhibit the following behaviors: a score of 0: no deficits; a score of 1: when the tail is lifted, the forelimb on the paralyzed side is retracted and flexed under the abdomen, while the normal side extends toward the surface; a score of 2: aside from the behavior of a score of 1, when lying prone on the surface, there is significantly less resistance to pushing objects toward the paralyzed side compared to the normal side; a score of 3: aside from the behaviors of a score of 1 and 2, the animal rotates toward the paralyzed side while walking.
The mNSS testWe evaluated the neurological deficits using mNSS as described previously [29]: a score of 1~6 indicated mild impairment; a score of 7 ~ 12 indicated moderate impairment; and a score of 13 ~ 18 indicated severe impairment. The mNSS comprised a motor, sensory, balance, and reflex test. In brief, the rats were placed on a customized balance beam (20 mm × 20 mm × 200 mm), fixed at a height of 10 cm above the table surface, allowing the animals to walk along the wooden beam. The performance of rats was observed and scored according to the table entries [24], with each rat assessed three times.
The cylinder testTo evaluate the motor functional recovery, a cylinder test was performed at 0, 1, 4, 7, and 14 days post-stroke [34]. In brief, the rats were acclimated in a customized transparent colorless acrylic cylinder measuring 400 mm (height) × 200 mm (outer diameter) × 190 mm (inner diameter) for 5 min. Their forelimb placements against the cylinder wall were then observed and recorded for a total of 20 trials for three independent times. The score was calculated as: (number of ipsilateral forelimb placements - number of contralateral forelimb placements) / (number of ipsilateral forelimb placements + number of contralateral forelimb placements + bilateral forelimb placements).
The adhesive removal testTo detect the sensorimotor function, an adhesive removal test was performed at 0, 1, 4, 7, and 14 days post-stroke [38]. The investigator was blinded to the experimental groups for the evaluation and the statistical analysis. In brief, the rats were allowed to acclimate in a clean, customized, transparent glass box (300 mm × 400 mm × 600 mm)for 2 min before testing. A piece of tape (30 mm × 40 mm) was then applied to the paw pad of the contralateral side of the rats. In a quiet environment, the rats were allowed to move freely, and the time taken to notice the tape and the time taken to remove it were observed and recorded three independent times.
Perfusion and tissue preparationFor tissue preparation, rats were anesthetized with 2% pentobarbital sodium via intraperitoneal injection. A thoracotomy was performed, and the animals were manually perfused with PBS through the left ventricle. The rats were then decapitated, and the ipsilateral side of the brain and the spleen were extracted for western blotting and flow cytometry. For frozen sections, rats were perfused with 4% paraformaldehyde (PFA) (Solarbio, USA) following perfused with PBS, and then the whole brain or the spleen was immersed in 4% PFA and stored at 4℃, followed by subsequent frozen slices.
Quantification of infarct volumeAccording to previous methods, we used 2,3,5-triphenyltetrazolium chloride (TTC) (Solarbio, China) staining to detect the quantification of infarct volume [39]. The fresh brains were quickly removed, frozen at − 20 °C, and sliced into 2 mm-thick sections. The slices were then stained with a 2% solution of TTC at 37 °C for 30 min in the dark. Each brain slice’s infarction area was measured using Image J analysis software. The infarct volumes were expressed (with correction for the edema) as a percentage of total hemispheres. Briefly, the volumes of the ipsilateral and contralateral hemispheres were counted, while the relative lesion volume was computed as follows: lesion volume=(area of contralateral hemisphere − area of normal region in the ipsilateral hemisphere)/area of contralateral hemisphere×100%.
Nissl stainingTo investigate the neuroprotective effects of rTMS and/or MSCs, Nissl staining was conducted to evaluate neuronal survival and death. The tissue was fixed in 4% paraformaldehyde (PFA) (Biosharp, China) at 4⁰C for 24 h. Sagittal brain Sect. (10 μm) were mounted on slides for Nissl staining [40]. Specifically, the frozen sections were removed from the − 20°C refrigerator and allowed to reach room temperature. They were then fixed with a tissue fixative for 15 min and rinsed with a gentle stream of water. The tissue sections were immersed in a toluidine blue staining solution (Servicebio, China) for 2–5 min for Nissl staining. Subsequently, they were washed with water, slightly differentiated with 0.1% glacial acetic acid (Aladdin, USA, A433223-500 ml), and finally washed with running water to terminate the reaction. The degree of differentiation was monitored under the microscope. After rinsing with running water, the sections were dried in an oven. The sections were immersed in clean xylene(Macklin, China, X823000-100 ml) for 10 min to achieve transparency, and then sealed with neutral gum. Nissl-stained images were captured using 20× microscopy. In interpreting the results, Nissl bodies in neurons appear as dark blue particles, while the nucleus is light blue and the background is also light blue. In normal, undamaged neurons, the Nissl bodies were large and numerous, and the nucleoli were prominent. In damaged neurons, the number of Nissl bodies may decrease or even disappear, and the cell spacing may increase.
Western blottingFollowing anesthetization and cardiac perfusion, brain tissue was promptly excised from the injured area. Peri-infarct region tissues of the brain or PC12 cells were collected and lysed in 1×RIPA lysis buffer(Solarbio, China, R0020), with pre-added protease inhibitor(1:100, Solarbio, China, P6730-1 ml) and phosphatase inhibitor(1:100, Solarbio, China, P1260-1 ml). After centrifugation at 13,000 × g for 15 min at 4 °C, we collected the supernatant as the protein lysate. Sonication lysis at 80% power for 1 min (15 s for cells), shaking the protein supernatant for 5 s and resting for 5 s. A Pierce™ BCA protein Assay kit (Thermo Fisher, 23225, USA) was utilized to quantify the protein concentration. Subsequently, protein samples were separated by SDS-PAGE (EpiZyme, China, PG113) and transferred to a 0.45 μm or 0.20 μm pore-sized polyvinylidenedifluoride (PVDF) membrane (Millipore, USA). The membranes were incubated in blocking buffer (Tris-buffered saline containing 5% skim milk powder, Biosharp, BS102-500 g, China) at room temperature for 1 h and then incubated with certain primary at 4 °C overnight. The following day, after being washed three times with Tris-buffered brine Tween20 (TBST) solution, the PVDF membranes were incubated with the corresponding secondary antibodies at room temperature for 60 min. The utilized primary and secondary antibodies are listed in Supplementary Table 2. Finally, an enhanced chemiluminescence kit (Biosharp, BL520B, China, ) was employed to detect the immunoreactive bands. To calculate the conjugation yield, we used gel band quantification by Image J software.
Immunofluorescence (IF)Sections of brain tissue were subjected to immunofluorescence staining as described below. We used immunofluorescent staining to investigate the localization and expression level of NeuN, GSDMD, Caspase-8, ASC, RIPK3, Iba1, CD68, CD45, CD169, and TNF-α. Briefly, brain slices were attached to the glass slides, and PC12 cells were seeded and cultured on a 15 mm round coverslip (#801007, NEST, China) in 12-well plates. Brain slices or coverslips of PC12 cells were washed with PBS for 10 min, followed by fixation for 10 min with 4% PFA. Subsequently, Brain slices or coverslips of PC12 cells were permeabilized in 0.1% Triton X-100 for 10 min, followed by incubation with 3% BSA buffer (Sigma-Aldrich, V900933-100G, USA) for 60 min at room temperature. Finally, brain slices or coverslips of PC12 cells were incubated with corresponding primary and secondary antibodies in the dark (listed in Supplementary Table 2). Nuclei visualization was subjected to DAPI (#D9542, Sigma) staining for 3 min at room temperature. Images were acquired under fluorescence microscopy or using a Leica confocal microscope. The mean fluorescence intensity was calculated by image J.
Immunohistochemical (IHC) stainingBrain slices of each group were fixed using transcardial perfusion and immersion in 4% PFA for 20 min. Brain slices were washed with PBS three times (5 min per time), following drying at 56 °C for 20 min. Following antigen repair with EDTA(ZS, ZLI-9067, China), brain slices were washed with PBS three times (5 min per time). The standard streptavidin-biotin-peroxidase complex was used in IHC staining. H2O2 (0.3%) solution was used to block endogenous peroxidase activity. The brain slides were incubated with primary antibodies overnight at 4 °C. After washing with PBS three times, the brain sections were incubated with horseradish peroxidase-conjugated secondary antibodies( Servisbio, GB23302, China) for 30 min at room temperature. The utilized primary antibodies are listed in Supplementary Table 2. A kit (Dako REAL™ EnVision™ Detection System, Peroxidase/DAB+, Rabbit/Mouse, #K5007) was used to amplify the staining. Subsequently, brain slices were counterstained with hematoxylin, dehydrated, and visualized by a bright field microscope (E100, Nikon, Japan). The percentage number of Ly6G-positive was determined in high-power fields (200×) of each brain slice. Images were analyzed using ImageJ.
TUNEL assayAfter immunofluorescence staining of NeuN according to the procedure mentioned above, we used a TUNEL staining kit (Beyotime, C1088, China) to continue co-staining with NeuN to detect the apoptosis of neurons according to the instructions of the kit. In brief, after staining with the NeuN primary antibody and the corresponding secondary antibody, 100 µL of TUNEL equilibration buffer was first added to each slice and incubated for 5 min. Second, the equilibration buffer was discarded. Then, 50 µL of TUNEL reaction mixture containing 1 µL of TdT enzyme was added to each slice. Third, the slices were placed flat in a humid chamber and incubated in the dark at 37 °C for 2 h. Fourth, the reaction mixture was removed, and the slices were rinsed twice in a 1× PBS staining bath, each for 5 min. Next, the slices were washed three times with a buffer containing 0.1% Triton X-100 prepared in PBS, which contained 5 mg/mL BSA, each wash lasting 5 min to reduce background staining. Fifth, 2 µg/mL DAPI staining solution was added dropwise to each slice and incubated in the dark at room temperature for 10 min. After staining, the DAPI solution was gently removed, and the samples were rinsed three times in 1× PBS, each for 5 min. Finally, the samples were observed and analyzed using a Leica confocal microscope.
Enzyme-linked immunosorbent assay (ELISA) of cytokines in CSF and serumRats were anesthetized by intraperitoneal injection of 2% pentobarbital sodium (50 mg/kg). After reperfusion with pre-cooled saline flush, the abdominal cavity was opened layer by layer to isolate and expose the abdominal aorta. An appropriate amount of blood was withdrawn with a 10 mL syringe. The blood was naturally coagulated at room temperature for 10 ~ 20 min, centrifuged for 10 min (2000 rpm), and the supernatant was collected. Cerebrospinal fluid was obtained according to previous work [24]. IL-1β (YJ730206, MIbio, China), IL-6 (YJ730219, MIbio, China), and TNF-α (SU-b31063, MIbio, China) ELISA kits were used to determine the OD values of each cytokine in the serum and cerebrospinal fluid (CSF) samples according to the instructions of each kit. The linear regression equation of the standard curve was used to calculate the sample concentration.
Isolation and analysis of CFSE+ MSCs in the brainTo track the MSCs in the brain following transplantation via caudal veil, suspension of MSCs was reacted with 5,6- carboxyfluorescein diacetate, succinimidyl ester, hereafter referred to collectively as CFSE(ThermoFisher, C34570, USA). Briefly, sixth passage human umbilical cord mesenchymal stem cells (MSCs) cultured in serum-free medium were digested with Accutase at 37 °C. The cells were then resuspended in 1 mL of PBS, to which 1 µL of CFSE was added. The mixture was allowed to react for 20 min at room temperature, with gentle agitation during the staining period. Subsequently, the cells were briefly washed with serum-free medium to remove any residual dye from the quenching solution, then centrifuged at 800 rpm for 5 min to obtain a cell pellet. The pellet was washed once with PBS (GIBCO, USA) and diluted to a concentration of 4 × 10^6/mL in PBS to prepare the suspension for caudal vein injection.
To analyze CFSE + MSCs in the brain, mononuclear cells were isolated from brain tissue. Total cells were extracted using Percoll’s isolation method and subsequently analyzed by flow cytometry. Briefly, rats were anesthetized via intraperitoneal injection of 2% pentobarbital sodium (50 mg/kg) and then perfused through the left ventricle with pre-cooled 1 × HBSS (GIBCO, USA) until the liver appeared pale. The brain tissue was immediately removed and placed in pre-cooled RPMI 1640 medium, where surface impurities were eliminated. The tissue was thoroughly homogenized using a cell screen with a 70 μm aperture, and the resulting homogenate was centrifuged at 2,000 rpm for 10 min at 4 °C. Following the removal of the supernatant, the cells were resuspended in 7 mL of RPMI 1640 medium at room temperature. To create a 30% Percoll separation solution, 3 mL of 100% Percoll was added and mixed with the cell suspension. Next, 3 mL of 70% Percoll separation solution was added to a new 15 mL centrifuge tube. The 30% Percoll solution was then carefully layered on top of the 70% Percoll solution using a straw. The mixture was centrifuged at 2,000 rpm (with acceleration set to 3 and deceleration set to 2) for 30 min at 22 °C. After centrifugation, a distinct white layer of cells was observed between the 30% and 70% Percoll solutions, representing the mononuclear cells. Approximately 2–3 mL of this white cell layer was carefully transferred to a new centrifuge tube. Then, 10 mL of 1 × HBSS (containing 2% fetal bovine serum) was added, and the mixture was centrifuged at 2,000 rpm for 10 min at 4 °C. The cells were washed three times with 1 × HBSS. The isolated mononuclear cells were then resuspended in 1 × HBSS for flow cytometry analysis to determine the proportion of CFSE + cells (FITC channel). Flow cytometry was performed using a BD LSR flow cytometer, and the data were subsequently analyzed using FlowJo software.
In vitro oxygen-glucose deprivation/reoxygenation (OGD/R) model establishment and treatmentTo investigate the effects of rTMS or/and MSCs in vitro, we used an oxygen-glucose deprivation/reoxygenation (OGD/R) model and a transwell co-culture model. The OGD 4 h/R 24 h model mimicked the cerebral ischemia-reperfusion damage in vitro. Briefly, when PC12 in 12-well plates reached 60% confluence, the 90% 1640 medium(added 10% FBS, 100 IU/ml penicillin, and 100 µg/ml streptomycin) was replaced into a glucose-free 1640 medium(Servicebio, G4538, China)and incubated in a 1% hypoxic incubator (ThermoFisher Technologies, USA) for 4 h at 37 °C. After 4 h of oxygen-glucose deprivation, the glucose-free 1640 medium was replaced into 90% 1640 medium (GIBCO, USA) supplemented with 10% FBS, 100 IU/mL penicillin, and 100 µg/mL streptomycin at 37 °C in a humidified incubator with 5% CO2 and 95% air for 24 h. The OGD/R model was successfully established.
For the treatment regimen, cells were subjected to 5 groups: PC12 cells without any intervention (control); PC12 cells suffered from OGD 4 h/R 24 h(OGDR) once the cells reached 60%~70% confluence; PC12 cells suffered from OGDR followed by rTMS treatment for every 8 h (OGDR + rTMS); PC12 cells suffer from OGDR followed by co-cultured with MSCs for 48 h (OGDR + MSCs); PC12 cells suffer from OGDR followed by co-cultured with MSCs for 48 h and rTMS treatment for every 8 h (OGDR + MSCs + rTMS). The co-culture between PC12 cells and MSCs was conducted by a transwell co-culture system.
Transwell co-culture systemPC12 cells (1 × 10^5 cells per well) were seeded in 12-well plates in 90% 1640 medium supplemented with 10% FBS, 100 IU/ml penicillin, and 100 µg/ml streptomycin at 37 °C in a humidified incubator with 5% CO2 and 95% air, and subjected to OGD/R modeling or control treatment when cells reached 60% confluence. At the onset of reoxygenation, a 4 µm Transwell chamber (size:12-well, Coning, #3401, USA) was placed atop the well plates, and MSCs suspension in a serum-free medium (2 × 10^5 cells per well) were seeded in the upper chamber. We subsequently put the upper chamber upon the 12-well plates with OGD/R modeling or control-treated PC12 cells as the lower chamber. Following 24 h of incubation, the cell suspensions from the 12-well plates were collected for western blotting and flow cytometry.
Apoptosis detection by flow cytometryAnnexin V-PI kit (A211-01, Vazyme, China) was used to detect the apoptosis of PC12 cells. According to the kit’s instructions, PC12 cells in each group were first detached from 12-well plates by incubating with 0.25% trypsin (without EDTA) for 1 ~ 2 min at 37 °C after treatment for 48 h. About 5 × 10^5 cells per well were collected, centrifuged at 1800 rpm for 5 min at 4 °C, and removed the liquid supernatant. Next, the cells were washed by pre-cooling PBS twice at 1800 rpm for 5 min at 4 °C. Then, cells were suspended in 100 µL 1× Binding Buffer. Finally, we added 5 µL Annexin -FITC and 5 µL PI staining solution into the cell suspension, incubated them for 15 min in the dark at room temperature, and detected the samples by flow cytometry within 1 h. All flow cytometric analyses were conducted with Gallios (Beckman Coulter) flow cytometers, and the data were analysed using the Kaluza (Beckman Coulter) software packages.
Cell viability assayCell viability was measured using a Cell Counting Kit-8 (CCK-8) (Dojindo, Kumamoto, Japan). When the PC12 cells were adherently cultured, PC12 cells(10^4 cells per well) were seeded in 96-well plates and cultured in 90% 1640 medium supplemented with 10% FBS, 100 IU/ml penicillin, and 100 µg/ml streptomycin at 37 °C in a humidified incubator with 5% CO2 and 95% air. When PC12 cells reached 60% confluence, PC12 cells in the MSCs group and MSCs + rTMS group were cultured with the medium supernatant of MSCs for another 48 h. Then, the medium was replaced with fresh medium containing 10 µl CCK8 reagent. Then plates were incubated for 2 h at 37 °C. Relative cell numbers were calculated by measuring the absorbance of each well at 450 nm using a multifunctional microplate reader (Spark 10 M, Switzerland).
RNA sequencing and bioinformatics analysisTo compare the gene profiles of the PC12 neurons, RNA sequencing was carried out in triplicate. Using TRIzol reagent and following the manufacturer’s instructions (Invitrogen, USA), total RNA was isolated from PC12 neurons in the OGDR group, OGDR + MSCs group, and the OGDR + MSCs + rTMS group. We measured the concentrations of the entire RNA sample using a NanoDrop ND-2000 instrument. Illumina NovaSeq 6000 platform (Annoroad Gene Technology, Co., Ltd, Beijing, China) was used for RNA sequencing. The original fastq files were then processed with fastp(version 0.2.0) for quality control in order to remove the low quality reads and adapter. After quality control, reads were mapped to rat genome reference (version Rattus_norvegicus.mRatBN7.2) using Hisat2 (version 2.1.0). The Htseq-count(version 0.11.2) were used to generate the gene count matrix. DESeq2(version 1.40.1) were used to perform differential gene analysis. A P value < 0.05 and absolute log2FoldChange value above 1 was considered statistically significant. The online database genemania (https://genemania.org/) and of trrust (and https://www.grnpedia.org/trrust/) were used to analyze protein-protein interaction (PPI) networks.
Statistical analysisStatistical analysis and plotting were performed using GraphPad Prism 9.0 software. All data were presented as the mean ± S.E.M. from at least three independent experiments. Result of infarction volume and nerve function scores were analyzed by Two-Way ANOVA, ELISA results were analyzed by One-Way ANOVA, and qPCR results were analyzed by T-test. P < 0.05 was considered statistically significant.
留言 (0)