
記住我
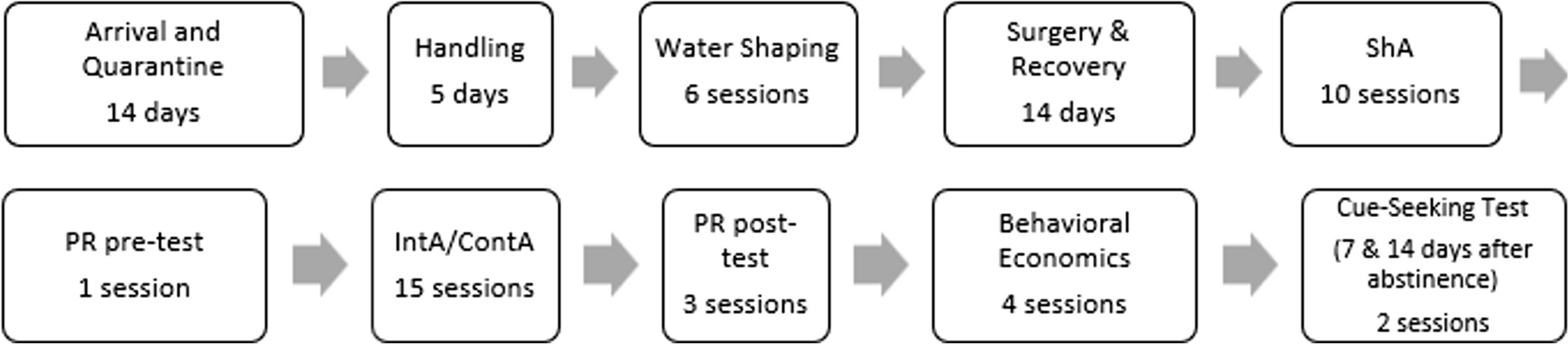







An overview of study design is provided in Fig. 1.
Fig. 1
This was an open-label, randomized, 5-period, ten-sequence, single use, cross-over study conducted at a single site (Clinical Trial Consultants AB, Uppsala, Sweden). The study included a screening visit, five product use and assessment visits, and a follow-up phone call 6–8 days after the last study product use. Product use visits were separated by washout periods of at least 24 h between the start of each product use to avoid carryover effects. Screening occurred within 28 days prior to the first product use and consisted of an eligibility assessment, review of health status and nicotine/tobacco use history. Baseline characteristics of the participants, such as gender, age, body mass index (BMI), their daily consumption of snus and nicotine content of their usual brand of snus, were collected at screening.
Participants were randomly assigned to one of ten sequences, balanced with three participants per sequence, according to a computer-generated randomization list, prior to the first product use. The original randomization list was kept by the randomizer. The use of any nicotine-containing products aside from the study products was not allowed from 12 h before the start of each product use and until the participants left the study site. Additionally, participants were not allowed to change their usual brand of snus during the study. At each product use and assessment visit, after 30 min of use (see Study products below), the used Nordic Spirit and LD snus pouches were collected and frozen at −20 °C within 5 min for later measurement of residual nicotine. Participants were instructed not to eat, drink, chew gum or brush their teeth from 30 min before product use, during product use and until 30 min after the end of product use. Blood samples for PK analysis were collected at pre-determined time points from pre-use up to 8 h after the start of product use. Vital signs, 12-lead electrocardiograms (ECG) and self-assessment of subjective product experience were also recorded at pre-determined time points. End of product use safety assessments (see Study assessments below) were performed after the last PK blood sampling during the last visit to the clinical site.
ParticipantsHealthy male and female Swedish portion snus users aged 19–64 years with a BMI ≥ 18.5 and ≤ 30.0 kg/m2 were eligible for study participation. Participants were exclusive and daily (7 days per week) users of Swedish snus with nicotine contents ≥ 8 mg/pouch for > 6 months prior to screening (single, occasional smoking of conventional cigarettes within 14 days prior to screening was allowed) and had urine cotinine levels > 200 ng/mL at screening. Prospective participants who planned to quit nicotine/tobacco use during the study period, or who would postpone an attempt to quit to participate in the study were excluded. Full details of eligibility criteria are available from https://doi.org/10.1186/ISRCTN75583947. Study participants were informed that they were free to quit nicotine/tobacco use and withdraw from the study at any time, for any reason, without forfeiting the right to appropriate follow-up. In total, 67 prospective participants were screened, and 30 were enrolled in the study.
Study productsAll study products were commercially available in Sweden at the time of the study. The products evaluated were: (a) Nordic Spirit mint-flavored NPs containing 6 mg nicotine/pouch; (b) Nordic Spirit mint-flavored NPs containing 9 mg nicotine/pouch; (c) Nordic Spirit spearmint-flavored NPs containing 11.2 mg nicotine/pouch; (d) conventional Swedish pre-portioned snus, bergamot-flavored LD Original Vit Stark portion containing 11.2 mg nicotine/portion; and (e) an NRT mint-flavored nicotine gum, Nicorette®, containing 4 mg nicotine/unit. Nordic Spirit NPs and LD snus were manufactured by Nordic Snus AB (a JT International SA company) and Nicorette® gum was manufactured by McNeil AB.
Study participants used each of the five products, in a randomized order, on separate days. the participants were instructed to place the Nordic Spirit NPs and LD snus between the upper lip and gum and to keep it in position for 30 min. For the Nicorette® gum, participants were instructed to chew the gum slowly ad libitum for 30 min.
Study assessmentsPharmacokinetics (PK) analysesVenous blood samples were collected within 10 min prior to each study product use as well as at the following time points after the start of product use: 5, 8, 10, 15, 20, 30, and 45 min, as well as 1, 1.5, 2, 3, 4, 6, and 8 h. Separated plasma samples were analyzed for nicotine concentrations by Lablytica Life Science AB (Uppsala, Sweden) by means of a validated liquid chromatography-tandem mass spectrometry (LC–MS/MS) method. This method was validated according to “Guideline on bioanalytical method validation, European Medicines Agency, 2011 (21 July 2011, EMEA/CHMP/EWP/192217/2009, Committee for Medicinal Products for Human Use (CHMP). PK parameters were calculated using non-compartmental analysis using Phoenix WinNonlin® version 8.1 (Certara, Princeton NJ, United States). The maximum observed nicotine concentration in plasma (Cmax) and time to Cmax (Tmax) were derived from the observed plasma concentration data, while AUC (area under the plasma concentration vs. time curve) parameters AUC0–60 and AUC0-last were assessed by integration of the plasma concentration vs. time curve using linear interpolation for increasing plasma levels and logarithmic interpolation for decreasing plasma levels (linear up/log down method). PK parameters were calculated for both unadjusted and baseline-adjusted plasma concentration data. AUC0-last was calculated from time 0 to the time of the last detectable plasma concentration. AUC0–60 is the AUC truncated at 60 min. All calculations were based on actual sampling times recorded during the study. Concentrations below the lower limit of quantification (LLOQ) occurring before Cmax were treated as zero. Concentrations below LLOQ occurring after Cmax were omitted from the analysis.
Nicotine extractionThe residual nicotine content of the used Nordic Spirit and LD snus pouches was determined by Lablytica Life Science AB (Uppsala, Sweden) using ultra-performance liquid chromatography coupled with ultraviolet detection (UPLC-UV) after extraction using 1:1 methanol–water. This analytical method has been validated according to “Nonclinical Dose Formulation Analysis Method Validation and Sample Analysis, 2010 (4 December 2010, AAPS Journal)” and “A Global GLP approach to Formulation Analysis Method Validation and Sample Analysis, 2011 (ISSN 2153–2435 PAA). Unused Nordic Spirit and LD snus pouches served to determine baseline nicotine content. The difference between the residual nicotine content in the used pouches and the baseline nicotine content in the reference pouches was used to calculate the extracted nicotine amounts.
Subjective measures questionnairesAt the end of each 30 min product use session, the participants filled out a modified product evaluation scale (modified PES) paper questionnaire to subjectively report their product use experience. This questionnaire was composed of the items 1 to 12 of the PES (Product Evaluation Scale). The PES (Hatsukami et al. 2013) is a modified consumer reported outcome measures questionnaire for oral nicotine products adapted from the modified cigarette evaluation questionnaire (Cappelleri et al. 2007).
The modified PES questionnaire contained self-evaluation questions for five subscales covering Product Satisfaction (Was it satisfying?, Did it taste good?, Did you enjoy it?), Psychological reward (Did it calm you down?; Did it make you feel more awake?; Did it make you feel less irritable?; Did it help you concentrate?; Did it reduce your hunger for food?), Aversion (Did it make you dizzy?; Did it make you nauseous?), Craving reduction (Did it immediately relieve your craving for a tobacco product?) and Enjoyment of mouth sensations (Did you enjoy the sensations in your mouth?). Each question was rated on a 7-point graded scale where one corresponded to “not at all” and seven to “extremely”.
A visual analog scale (VAS) questionnaire was used to assess the intent to use the product again 8 h after the start of each product use. The question “If given the opportunity, would you use this product again?” was answered with a 100 mm VAS scale printed on a sheet of paper anchored with “Definitely would not” at 0 mm and “Definitely would” at 100 mm. The participants placed a vertical mark anywhere on the 0–100 mm line to reflect their intent to use the product again.
Safety assessmentsAdverse events (AEs), including serious adverse events (SAEs), self-reported by the participants, observed by the study medical personnel, or elicited from the participants based on non-leading questions, were collected from the first use of study product until the end of the study. An AE was defined as any untoward medical occurrence in a study participant that used a study product, and which did not necessarily have a causal relationship with this study product (European Medicines Agency 1995). AEs were graded for severity (grade 1 to grade 5) following the common terminology criteria for AEs (CTCAE) v5.0 and assessed as unlikely, possibly, or probably related to the study products. Vital signs (systolic/diastolic blood pressure and pulse rate) were measured prior to each product use as well as at 5 min, 30 min and 1 h after the start of product use. 12-lead electrocardiograms (ECGs) were recorded prior to each product use. Safety assessments carried out after the last product use and prior to discharge from the clinical site included vital signs and ECG measurements, physical examination, blood and urine sampling for standard care clinical chemistry and hematology laboratory parameters, and the collection of AEs and uses of concomitant medication.
Statistical analysesThe number of study participants was considered sufficient for this exploratory study based on similar study designs published in the literature (Rensch et al. 2021; Liu et al. 2022; Chapman et al. 2022). Descriptive summaries and statistical analyses were performed using SAS Version 9.4 (SAS Institute, Inc., Cary NC, United States). Statistical comparisons of each PK parameter were made between each of the Nordic Spirit NPs and the two comparator products. Log transformed nicotine Cmax and AUC0-last estimates were evaluated separately in a linear mixed-effects analysis of variance (ANOVA) model with fixed effects for period, sequence, and product, and a random effect for participant. The above product differences were back transformed to present the ratios of geometric least squares (LS) means and 95% confidence intervals (CIs) of each test product versus each comparator product from the same model. The nicotine Tmax was analyzed separately for each of the above-mentioned product comparisons using a Wilcoxon signed-rank test. Estimates of the median difference based on the observed medians, 95% CIs, and p-values from the Wilcoxon signed-rank test were calculated. Linear regression analysis was performed for assessing the relationship between the extracted amount of nicotine and PK parameters (Cmax and AUC) with calculating Pearson correlation coefficient and the p value.
留言 (0)