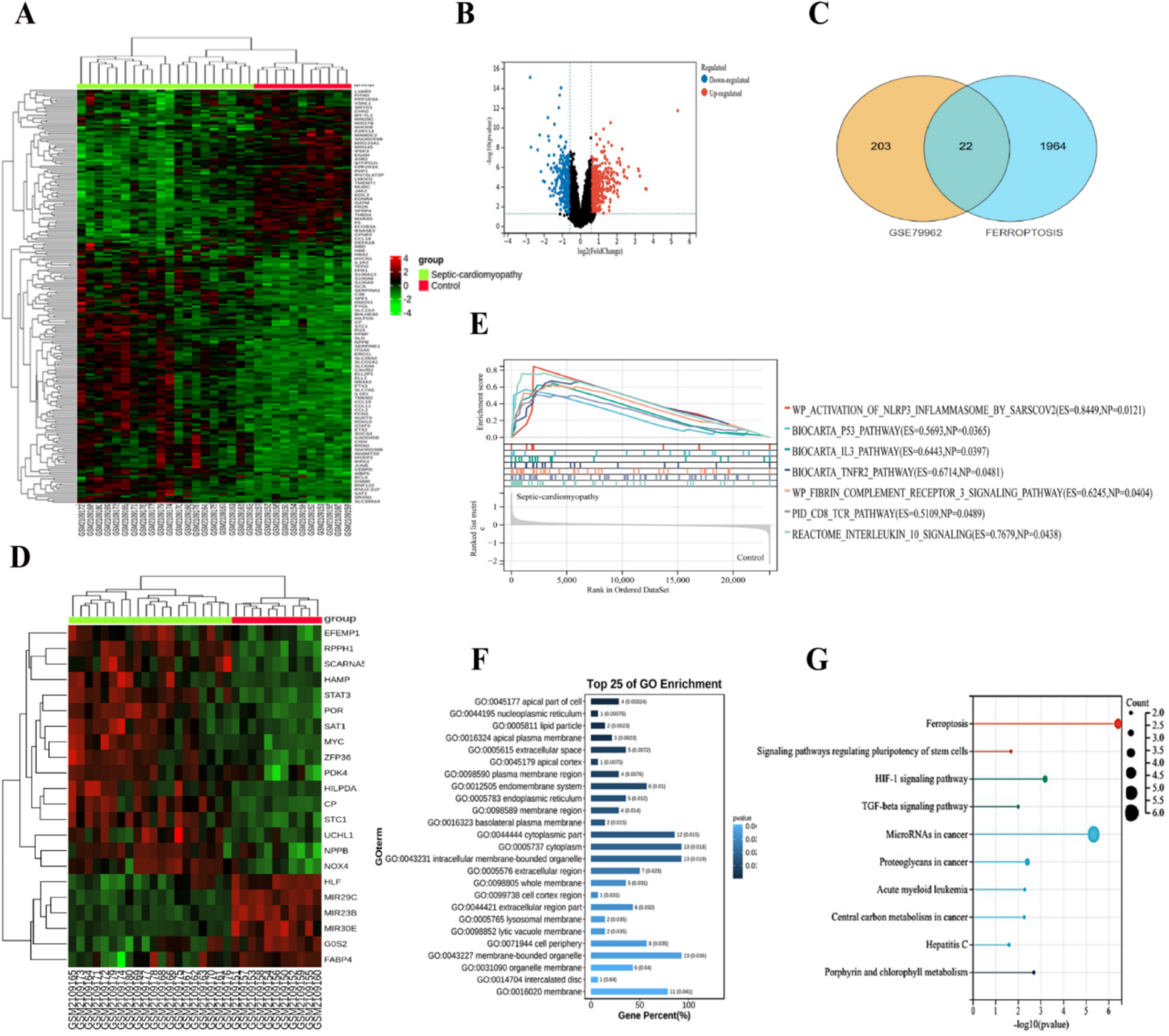
Study population and DNA extraction
In this study, we enrolled 40 patients diagnosed with TOF-PS and 60 patients diagnosed with pulmonary atresia with ventricular septal defect (PA-VSD), confirmed by both cardiac echocardiography and evaluation performed by a specialist cardiologist. Additionally, 120 unrelated healthy controls without congenital heart valve defects were recruited from Shanghai Xin Hua Hospital. All participants in the patient cohort were members of the Chinese Han community, ranging in age from newborns to 17 years. DNA extraction was performed according to the standard protocol, employing the QIAamp DNA Blood Mini Kit (QIAGEN, Germany). The study design adhered to the principles of the Declaration of Helsinki.
WES, Sanger sequencing, and variant analysis
WES was conducted on all samples utilizing the Illumina HiSeq 2500 platform to screen for mutations. Nonsynonymous variations in the encoding regions of MFNG, with a frequency of less than 0.1% in normal control individuals (i.e., less than 0.1% in the 120 healthy participants and publicly available variation databases, including ExAC), were considered. To further validate the candidate MFNG variant, Sanger sequencing was employed. In this study, rare mutations were defined as those with a minor allele frequency (MAF) less than 0.5%. The pathogenicity of the MFNG mutation was assessed using various bioinformatic websites, including SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html), Mutation Taster (http://www.mutationtaster.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and CADD (https://cadd.gs.washington.edu/info).
Conservation analysis of MFNG protein sequence
MFNG protein sequences from various species, including Homo sapiens (human), Mus musculus (house mouse), Rattus norvegicus (rat), Macaca mulatta (rhesus monkey), Bos taurus (cattle), and Pan troglodytes (chimpanzee), were obtained from NCBI (https://www.ncbi.nlm.nih.gov/protein/). The Clustal X software (http://www.clustal.org) was utilized to assess the conservation of MFNG protein sequences.
Plasmids and site-specific mutagenesis
A full-length human MFNG ORF was inserted into the pCDH-CMV-MCS-EF1-copGFP-T2A-Puro vector to construct the wild-type (WT) MFNG expression plasmid. To introduce mutations, the QuikChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) was employed. All plasmids were subjected to verification for integrity and accuracy via Sanger sequencing.
Cell culture, transient transfection, and treatment
Human umbilical endothelial cells (HUVECs) from passages four to seven were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen, CA, USA), supplemented with 10% fetal bovine serum (MP Biomedicals, USA) and 1% penicillin–streptomycin solution (Gibco, USA). Transfections were performed by separately introducing 3 µg of empty vector, oe-MFNG, and oe-MUT plasmids into HUVECs using FuGene HD (Promega, USA) when the cell concentration in the 6-well plate reached 70–80%. After 24 or 48 h of transient transfection, HUVECs were harvested for subsequent experiments. To induce EndMT in vitro, HUVECs were treated with transforming growth factor beta 2 (TGFβ2, 10 ng/ml, R&D, 302-B2) for 48 h as the treatment group, or with 0.1% bovine serum albumin (BSA) for 48 h as the control group when they reached 70–80% confluence. HUVECs were starved with basal medium for 12–16 h before treatment, and the medium with TGFβ2 or 0.1% BSA was freshly replaced every day. Additionally, DAPT (Sigma-Aldrich, St. Louis, CA, USA) was added to the cell medium (20 µg/ml) and renewed every other day to inhibit the Notch pathway.
Lentivirus assembly and transduction
To stably overexpress MFNG in HUVECs, the pCDH vector containing human MFNG (pCDH-MFNG) was constructed. After synthesizing the MFNG overexpression lentiviral vector, a mixture of psPAX2, pMD2.G, and the modified pCDH-CMV-MCS-EF1-copGFP-T2A-Puro containing the MFNG coding sequence was co-transfected into Hek-293 T cells at a ratio of 4:2:1. Polyethyleneimine (PEI) was used for transfection at a ratio of 3:1 (PEI µg: total DNA µg). The viral supernatants were collected at 48 and 72 h and filtered through a 0.45-µm filter. Subsequently, HUVECs were transduced with the lentivirus at a 1:1 ratio of viral supernatants to fresh medium, supplemented with 8 µg/ml polybrene. After 48 h of incubation, 2 µg/ml puromycin was added to the culture medium to select for transfected cells. To knock down MFNG in HUVECs, recombinant lentiviruses carrying MFNG-targeting small interfering RNA (sh-MFNG) were designed and synthesized. The lentiviral transfer vector (pLKO.1-TRC) was constructed, and sh-MFNG expression was driven by the U6 promoter. Lentivirus packaging and cell transfection followed previously established protocols.
Wound healing assay
HUVECs were seeded into 6-well plates at a density of 5 × 105 cells per well. When the cells reached approximately 95% confluence, they were serum-starved for 16 h. Subsequently, cell monolayers were scratched using 200-µl pipette tips and washed three times with PBS. The cells were then incubated with fresh medium containing 1% FBS for 24 h, and images were captured at 0 and 24 h after scratching. ImageJ software was used to analyze the wound area healing.
Transwell assay
Samples with 2 × 105 cells were resuspended in a serum-free culture medium after trypsinization and plated on the upper layer of transwell chambers (Corning, USA). The residue cells on the upper side migrated toward the lower chamber with a gradient of 20% FBS. The cells on the top surface of the membrane were wiped out, leaving only the cells with the ability to migrate. These cells were washed three times with PBS, fixed with 4% paraformaldehyde (PFA) for 15 min, and stained with crystal violet for 30 min after 24 h of incubation. The inverted microscope was used to capture the images of migrated cells.
Quantitative real-time PCR
Total RNA was extracted from HUVECs and reverse transcribed into cDNA using a reverse transcriptase kit (TaKaRa, Japan). Quantitative real-time PCR (qRT-PCR) was performed on the Applied Biosystems 7500 system using SYBR Green (TaKaRa, Japan). GAPDH was used as an internal reference. The qRT-PCR settings were as follows: 3 min at 50 °C and 30 s at 95 °C, followed by 40 cycles of 10 s at 95 °C and 30 s at 60 °C. The relative gene expression was quantified using the ΔΔCt calculation method. The specific primer sequences are listed in supplementary Table 1.
Western blot
HUVECs were collected by scraping and lysed with RIPA buffer (Beyotime, P0013B) containing a mixture of protease and phosphatase inhibitors (PMSF, 1:100) for 30 min on ice. Protein concentration was determined using the BCA protein quantification kit (Beyotime, P0010). Equal amounts of protein from each group were separated by SDS-PAGE and transferred to an NC membrane. After blocking with 5% skim milk for 1.5 h at room temperature, the membranes were incubated overnight at 4 °C with specific primary antibodies. Subsequently, the membranes were incubated with secondary antibodies conjugated to horseradish peroxidase (HRP) for 1.5 h at room temperature. Gel images were captured using the Tanon gel imaging system, and each band was normalized to the loading control β-actin. Immunoblots were analyzed by densitometry using ImageJ software.
Cellular and tissue immunofluorescence staining
For cellular staining, HUVECs cultured on coverslips were fixed in 4% PFA for 15 min. Subsequently, they were permeabilized with 0.5% Triton X-100 in PBS for 10 min and then blocked with 5% BSA for 1.5 h at room temperature. Following the removal of the blocking buffer, the primary antibody diluted in the blocking buffer was applied to the HUVECs and incubated overnight at 4 °C. The next day, HUVECs were exposed to fluorescence-conjugated secondary antibodies for 1.5 h at room temperature to visualize the specific target protein. After the secondary antibody incubation, DAPI (Vector Laboratories, USA) with an anti-fluorescence quenching agent was used to stain cell nuclei. For staining of tissue sections, paraffin-embedded heart tissue sections were dewaxed in xylene and rehydrated through a graded alcohol series: 100% ethanol, 90% ethanol, 75% ethanol, and distilled water. Antigen retrieval was performed by heating in sodium citrate buffer (pH = 6) for 6 min, followed by cooling to room temperature. The steps for permeabilization of tissue sections, blocking with 5% BSA, and subsequent incubation with primary and secondary antibodies were the same as those described for cells. Finally, a fluorescence microscope was used to detect red, green, and blue channels.
Maintenance of zebrafish, microinjection of morpholinos into the embryos, and hematoxylin–eosin staining
The Tg (myl7: EGFP) transgenic line embryos labelled with green fluorescence were purchased from the China Zebrafish Resource Center and maintained at 28.5 °C under standard laboratory conditions. All the experiments received approval from the Animal Care and Use Committee of Xinhua Hospital. Morpholino antisense oligos (MO), chemically modified oligonucleotides, can cause aberrant splicing by targeting splice junctions. They remain effective in blocking protein synthesis of the targeted gene(s) for 3–4 days. Zebrafish MFNG MO1 (5′-AGCACCATTAGCTTACTCACTTTTT-3′) and MO2 (5′-ATGAATAGATGTGATACTGACCGTC-3′) were designed and synthesized by Gene Tools (OR, USA). A standard control MO (5ʹ-CCTCTTACCTCAGTTACAATTTATA-3ʹ) was employed as a negative control. For knockdown assays, MO were injected into the yolk sac of embryos when they were at the 1–2 cell stage using a pressure microinjection device (PICOSPRITZER® III, Parker, USA). qRT-PCR analysis of MFNG cDNA collected from the whole embryos 30 h after the injection was employed to confirm MO efficiency. The preliminary experiment was then carried out to evaluate the appropriate injection concentration of MO. The embryos were examined periodically, and their phenotypes within each group were recorded at 72 hpf. Zebrafish embryos were fixed in 4% PFA, followed by dehydration through a graded ethanol series, clearing in xylene, and embedding in paraffin wax. Longitudinal sections of 4 µm thickness were dewaxed, dried, and stained with hematoxylin and eosin (HE) according to standard protocols. The resulting images were acquired using a Leica microscope.
In vitro OFT explant culture and isolation of ECs from the OFT explants
The cultures of the OFT explants were carried out in accordance with the previously described methodology. The rat tail collagen-type I matrices (Corning, 354236) were allowed to polymerize in an incubator set at 37 °C and 5% CO2. The OFT tissue was dissected in sterile PBS from E10.5 embryos. The OFT cushions were exposed by using sharp tweezers and secured with the cushion-side facing downwards on the collagen gels, which were allowed to facilitate the attachment of the cushions. After overnight adhesion, adenoviral infections were performed to analyze the effects of MFNG overexpression and mutation on the OFT explants. To express MFNG or MFNG mutation, the OFT explants were infected with adenovirus containing either MFNG (AdV-MFNG) or the MFNG mutation (AdV-MUT), which were tagged with GFP, or empty adenovirus (AdV-GFP). Endocardial cells that underwent transformation were identified as the spindle-shaped cells that migrated away from the explants or invaded the gel. Bright-field images and GFP fluorescence images of OFT explants were obtained using a Leica microscope. Explants were fixed and stained with α-SMA to detect mesenchymal cells. Cell numbers were determined by manual counting. The ECs isolated from the OFT explants were collected for further research after infection with AdV-MFNG, AdV-MUT, or AdV-GFP and subsequent removal of the myocardial tissue using collagenase type II (Sigma-Aldrich).
Statistical analysis
All measurement data were analyzed using GraphPad Prism 8 and presented as the mean ± standard error (SEM). The experiments were independently repeated at least three times. The statistical significance of observed differences was assessed using a two-tailed t-test between the two groups. One-way analysis of variance (ANOVA) with Tukey’s multiple comparison test when more than two groups are compared. Two-way ANOVA was used to test for differences between groups with multiple factors. For zebrafish MO assays, the chi-squared test (and Fisher’s exact test) was applied to compare the proportions of zebrafish embryonic phenotypes between the ctrl-MO group and the MFNG-MO group. A p-value less than 0.05 was considered significant.








留言 (0)