Chemicals
Human dermal fibroblast was sourced from Clonetics (San Diego, CA). Fetal bovine serum (FBS), trypsin–EDTA, and penicillin–streptomycin were provided from Lonza (Walkersville, MD, USA). Dulbecco’s modified Eagle’s medium (DMEM), mannitol, D-glucose, umbelliferon, and all other regents not mentioned elsewhere were obtained from Sigma-Aldrich Chemical (St. Louis, MO, USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin (Ig)G, goat anti-mouse, and donkey anti-goat IgG were acquired from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Essentially, fatty acid-free bovine serum albumin (BSA) and skim milk were supplied by Becton Dickinson Company (Sparks, MD, USA). Mouse monoclonal antibodies of collagen I (clone 3G3, catalog no. sc-293182), fibronectin (clone 2775–8, catalog no. sc-69681), pro-collagen I (clone M-60, catalog no. sc-30136), matrix metalloproteinase (MMP)-2 (clone 2C1, catalog no. sc-13594), MMP-9 (clone E-11, catalog no. sc-393859), EVL (clone B-1, catalog no.sc-376943), Fascin-1 (clone 55 K-2, catalog no. sc-21743), VEGF (clone VG-1, catalog no. sc-53462), E-cadherin (clone 5F133, catalog no. sc-71007), Occludin (clone E-5, catalog no. sc-133256), Has-2 (clone C-5, catalog no. sc-365263), HYAL (clone 1D10, catalog no. sc-101340), and AQP-3 (clone F-1, catalog no. sc-518001) were supplied by Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal antibody of ZO-1 (catalog no. #61–7300) was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Antibody of mouse polyclonal HIF-1α (clone D2U3T, catalog no. 3716S) was purchased from Cell Signaling Technology (Denver, MA, USA). Mouse monoclonal β-actin (clone AC-15, catalog no. A1978) antibody was provided by Sigma-Aldrich Chemical. All primary antibodies were diluted at a 1:1000 ratio in 5% BSA.
Cell culture
Human dermal fibroblasts were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 mM glutamine at 37 °C in a humidified incubator containing 5% CO2. Cells were seeding at 90% confluence in all experiments. Fibroblasts were incubated in 5.5 mM normal glucose or 33 mM high glucose for a hyperglycemia environment. Non-toxic concentrations of 1–20 µM umbelliferone were added in media for 3 days. Cell viability or cytotoxicity was measured by assaying with MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltertrazolium bromide). Cells were incubated with 1 mg/ml MTT solution at 37 °C for 3 h, forming an insoluble purple formazan product that was dissolved in isopropanol prior to reading the absorbance in a microplate reader at 570 nm.
In vivo animal experiments
Male C57BL/6 J mice of 5 weeks of age (average weight 20 ± 0.5 g) were purchased from DooYeol Biotech (Seoul, Korea). To introduce diabetes, mice were subjected to daily intraperitoneal (IP) injections of streptozotocin (STZ) (Sigma-Aldrich Chemical, St. Louis, MO, USA) dissolved in sodium citrate buffer (pH 4.5, 25 mg/kg/day) for five consecutive days. The induction of diabetes was confirmed by a fasting blood glucose concentration after the fifth day of STZ injection, and mice were divided into three subgroups (n = 9–10 for each subgroup). The first group was fed a standard laboratory chow diet (DooYeol Biotech, Seoul, Korea) as a control. The STZ-treated mice fed a high-fat diet (HFD) were divided randomly into two groups based on fasting glucose checks. One group received water orally after STZ administration, while the other group received oral administration of umbelliferone at a dosage of 10 mg/kg body weight for 10 weeks, excluding the day of wound formation and the following 2 days. The content in fat percentage of the diets is 60% kcal% fat. Mice were kept on a 12-h light/dark cycle at 23 ± 1 °C with 50 ± 10% relative humidity under specific pathogen-free conditions, fed a standard laboratory chow diet (CJ Feed, Seoul, Korea), and were provided with water ad libitum at the animal facility of Hallym University. All experiments were approved by the Committee on Animal Experimentation of Hallym University and performed in compliance with the University’s Guidelines for the Care and Use of Laboratory Animals (hallym(2023–18)). No mice died, and no apparent signs of exhaustion were observed during the experimental period.
Full-thickness excisional wound model
Mice were anesthetized with 250 mg/kg concentration of Avertin via intraperitoneal injection. After anesthesia, the dorsal hair was shaved to fully expose the skin, which was then rinsed with 70% ethanol. Four full-thickness wounds were created on each mouse using a 6-mm-diameter biopsy punch under sterile conditions. To determine the wound closure rate, wound areas were evaluated on days 0, 4, 8, 12, 14, and 18 post-wounding. Photographs were taken, and the wound area was measured by tracking the edge of the wound using ImageJ image analysis software (NIH, Bethesda, MD, USA). The percentage of wound closure was expressed as the ratio of the remaining area of the wound to the original area of the wound over time. The percentage of residual scar rate was expressed by taking the ratio of the scar area to the original area of the wound.
Western blot analysis
Whole-cell lysates were prepared from fibroblasts (3.5 × 105 cells) and skin tissue extracts from mouse skin using RIPA buffer. The skin tissue extracts were obtained from the skin of mice that were grown until 10 weeks. After anesthesia, skin samples from mice were collected from the mice, immediately extracted, and snap-frozen for biochemical assays. For western blot analysis, each sample was homogenized in lysis buffer and centrifuged at 3000 rpm for 10 min to remove any insoluble material. The supernatant was then transferred to a clean tube. Protein concentration was determined using the Lowry protein assay, and 30–50 µg of protein was used for the experiments. Equal amounts of proteins from cell lysates and tissue extracts were separated by electrophoresis on 8–12% SDS-PAGE and then transferred onto a nitrocellulose membrane. Nonspecific binding was blocked with either 3% fatty acid-free bovine serum albumin (BSA) or 5% nonfat dry skim milk for 3 h. The membrane was subsequently incubated overnight at 4 °C with primary antibodies targeting specific proteins, followed by washing in a TBS-T buffer for 10 min. Afterward, the membrane was incubated for 1 h with secondary antibodies (goat anti-rabbit IgG, goat anti-mouse IgG, or donkey anti-goat IgG) conjugated to HRP. Each target protein level was assessed using immobilon western chemiluminescent horseradish peroxidase substrate (Millipore Corp.) and detected through chemiluminescence using a ChemiDoc (Cytiva). Additionally, incubation with a mouse monoclonal β-actin antibody was performed for comparative controls. We performed a quantitative analysis of the western blot results using densitometry. The band intensities were measured using ImageJ software (NIH, Bethesda, MD, USA) and normalized to the corresponding β-actin bands. The relative expression levels were calculated and statistically analyzed.
Immunostaining
Fibroblasts were fixed with 4% formaldehyde for 10 min and permeabilized by 0.1% Triton X-100 on ice. To blockade the unspecific protein binding, fibroblasts were treated with 5% bovine serum albumin (BSA) for 1 h. The primary antibody for collagen I was prepared by diluting it at a ratio of 1:100 in 5% BSA and incubated overnight at 4 °C. The secondary antibody, Cy3-conjugated anti-mouse IgG (Sigma-Aldrich Chemical, St. Louis, MO, USA), was diluted at a ratio of 1:500 in PBS and used for the cytochemical staining of fibroblasts for 30 min. Nuclear staining was performed using 4′,6-diamidino-2-phenylindole (DAPI), which was diluted at a ratio of 1:1000 in PBS. Slide was mounted in glycerol (Sigma-Aldrich Chemical, St. Louis, MO, USA). Images were taken using an inverted microscope system for the visualization of collagen I (Nikon, Tokyo, Japan). Fluorescence intensity of immunostained cells, using a Cy3-red dye, was measured with the ImageJ software (National Institutes of Health, Bethesda, MD, USA). The red channel of the images was analyzed directly. Regions of interest (ROIs) corresponding to individual cells were selected. The mean fluorescence intensity within each ROI was quantified, and background fluorescence was subtracted. The results were normalized to control samples to account for variability between experiments.
Cell scratch wound healing assay
To evaluate the impact of glucose on dermal fibroblast motility, as well as the potential effects of umbelliferone, an in vitro scratch wound assay was employed. In this assay, fibroblasts were seeded onto a 12-well plate and allowed to incubate for 24 h in media containing 10% FBS (fetal bovine serum). Once the cells reached confluency, a scratch was created horizontally in each well using a pipette tip. After the scratch was made, the injured cells were further incubated for an additional 24 h in culture media containing 33 mM high glucose media, with or without the presence of 1–20 µM umbelliferone. Images of scratch wounds were captured in 2–3 microscopic fields per well using a microscope equipped with a Nikon camera (Nikon, Tokyo, Japan). Quantification of wound closure was performed using ImageJ software (NIH, Bethesda, MD, USA) normalized with the wound size at day 0 as 100%.
Rhodamine phalloidin staining
Fibroblasts (0.7 × 105 cells) were fixed with 4% formaldehyde for 10 min and permeabilized with 0.1% Triton X-100 for 10 min on ice. For red cytochemical staining, the cells were treated with red-fluorescent rhodamine phalloidin (Thermo Fisher Scientific, Waltham, USA) for 30 min. Nuclear counterstaining was performed using 4′,6-diamidino-2-phenylindole (DAPI). Each slide was mounted with VectaMount mounting medium (Vector Laboratories, Burlingame, CA, USA).
Hematoxylin and eosin staining of skin tissue and measurement of epidermis/dermis thickness and adipocyte size
Paraffin-embedded skin specimens were cut into 8 µM thick sections, dewaxed and rehydrated, and stained with eosin reagent to assess and hematoxylin for counterstaining. The stained tissue slides were examined using a microscope equipped. The thickness of skin epidermis, dermis, and adipocyte size were analyzed by ImageJ software.
Masson and Gomori trichrome staining
For the histological analysis, the skin was obtained at the end of the experiments and fixed in 10% buffered formalin. The skin tissues were then embedded in paraffin, and sections of 8 µM thickness were prepared. These sections were de-paraffinized and stained with Masson and Gomori trichrome to visualize collagen fibers and muscle fibers under light microscopy. The stained tissue sections were examined using an inverted microscope system. The analysis of Masson and Gomori staining intensity was performed using ImageJ software. Digital images were uploaded to ImageJ, and a scale was set using digital micrometer gauge readings to convert pixel units to microns. The proportions of collagen fibers (blue areas), cytoplasm, and muscle (red areas) were determined by setting the appropriate color thresholds, following the methodology [17].
In vivo skin evaluation
Hydration and TEWL of the dorsal skin of the mice were measured with a Corneometer CM 825 (Courage and Khazaka, Köln, Germany), and SCH was measured using GPSkin (GPOWER Inc., Seoul, South Korea), respectively. Three independent measurements from the same area of skin were averaged for each value.
Statistical analysis
Data were analyzed using SPSS (SPSS Inc., Chicago, IL, USA), and a mean difference of p < 0.05 was considered statistically significant. Different letters in the figures indicate significant differences between various treatment groups at p < 0.05, as determined by one-way ANOVA. Once a significant difference was recognized, Tukey’s test was conducted as a post hoc analysis to compare the differences between the groups. All data are presented as mean ± SD.


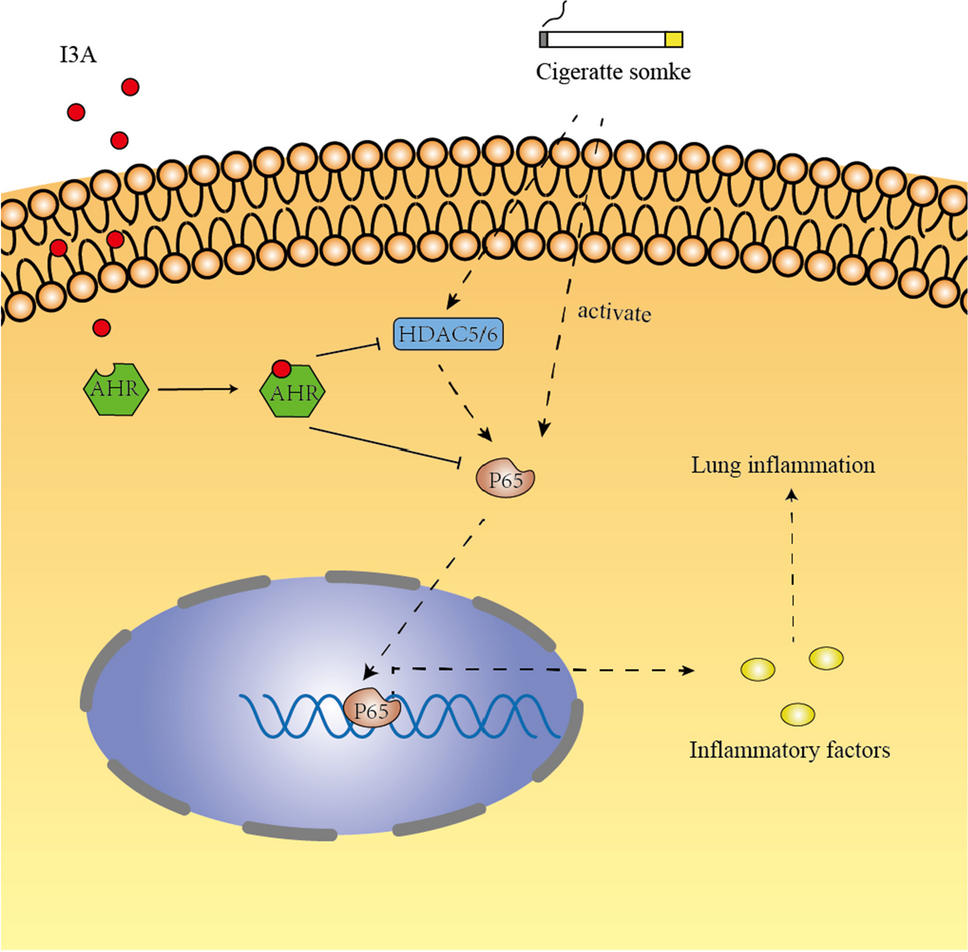
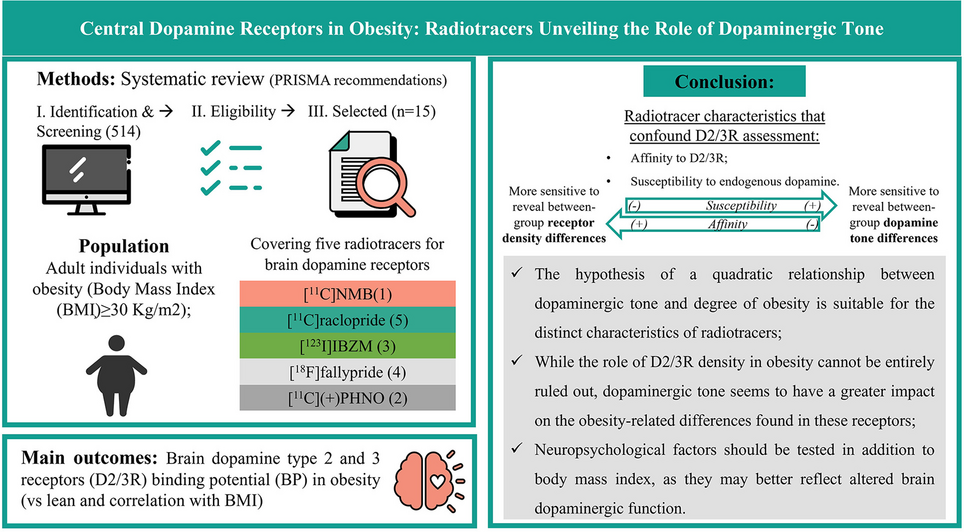
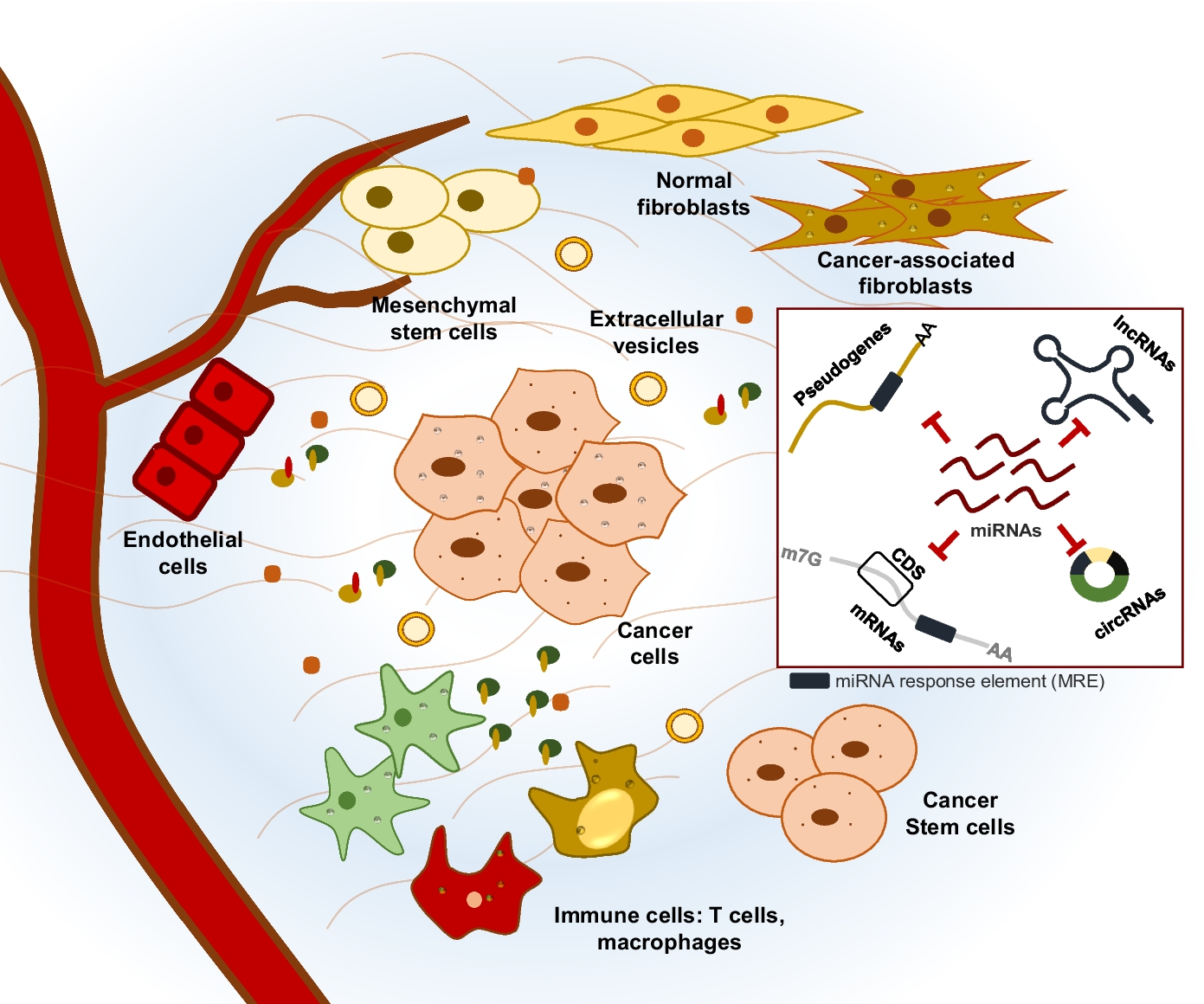
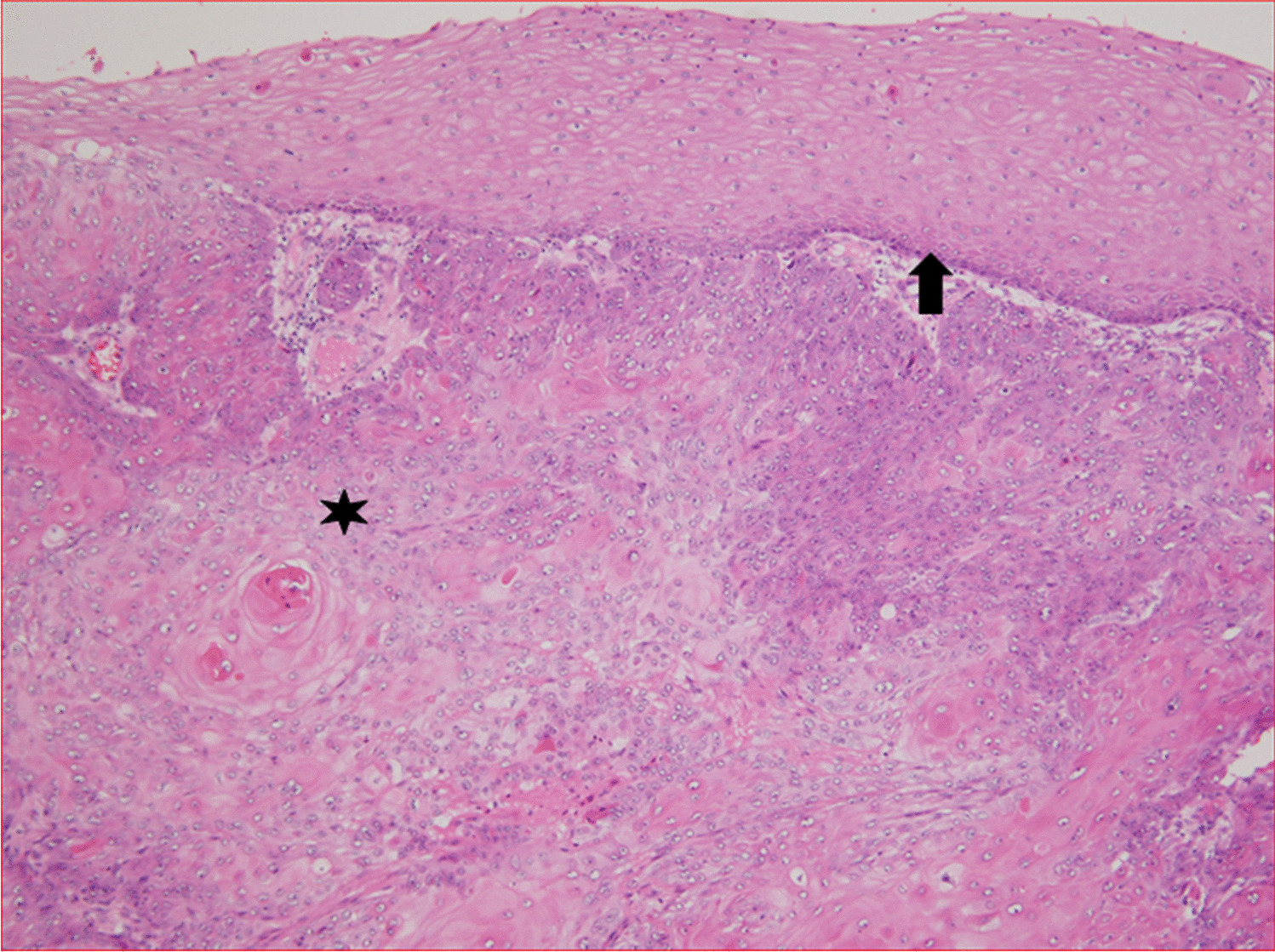



留言 (0)