Patient samples
Forty LUAD samples and adjacent non-tumorous lung tissues were collected from the Department of Thoracic Surgery at Lihuili Hospital, Ningbo Medical Center. All patients had not received treatment prior to surgery and were histopathologically diagnosed with LUAD postoperatively. The Ethics Committee of Lihuili Hospital, Ningbo Medical Center, approved this study and all patients provided informed consent (KY2022PJ140). This study does not involve the extent of blinding.
Cell culture
Human embryonic lung fibroblasts (MRC-5) were cultured in minimum essential medium (MEM; Gibco, MA, USA). The LUAD cell lines (HCC827, PC9, NCI-H1650, A549, NCI-H1975) were cultured in RPMI 1640 medium (Gibco). Both media were supplemented with 10% FBS (Pricella, Wuhan, China) and 1% penicillin/streptomycin (Beyotime, Shanghai, China). Routine mycoplasma detection and elimination were performed. Cell authentication was verified using short tandem repeat profiling.
Construction of osimertinib-resistant cell lines
The osimertinib-resistant cell line (H1975-OR) derived from NCI-H1975 cells was acquired from MeisenCTCC (Zhejiang, China). Resistance in the parental NCI-H1975 cell line was induced by gradually increasing osimertinib concentrations. Briefly, log-phase cells underwent shock induction using a gradient concentration increase method, starting with 500 nM osimertinib. The drug was removed after 24-h treatment, and the drug shock was repeated once the cells resumed growth. The drug concentration was increased by 500 nM every 15 days. The cells were continuously cultured for 6–9 months. The resistance index (RI) was calculated as the IC50 of the resistant cells divided by the parental cell IC50. An RI of 1–5 indicated low resistance, 5–15 indicated moderate resistance, and >15 indicated high resistance.
Construction of osimertinib-tolerant cells
H1975 osimertinib DTPCs (H1975-DTPCs) were generated by continuously exposing the parental H1975 cells to 1 µM osimertinib for 20 days. Subsequently, the cells were harvested, and their RNA and protein expression was analyzed using immunofluorescence, quantitative real-time PCR (qRT-PCR), and western blotting. The collected cells were then plated in 96-well plates for drug sensitivity testing.
Patient-derived organoid (PDO) construction and culture
LUAD organoids were derived from biopsy samples collected from patients at their initial diagnosis, prior to treatment. Additionally, osimertinib-resistant organoids were established from lung puncture tissues obtained from patients whose tumors had progressed despite osimertinib treatment. Upon excision, the tissues were immediately transported to a level 2 biosafety laboratory (BSL-2) for the primary organoid construction. The tissues were minced, digested with enzymatic solutions, and filtered to collect single cells, which were cultured in Matrigel (Corning, NY, USA). The organoid density and morphology were monitored daily, with fresh culture medium replaced every 2–3 days. The primary organoids typically formed solid or cystic structures measuring 50–100 µm within 2 weeks. The organoids could sustain ex vivo expansion and maintained stable morphology throughout the culture.
Lentiviral construction and infection and small interfering RNA (siRNA)
Lentiviral systems for gene overexpression or knockdown were purchased from GeneChem (Shanghai, China), with the lentiviral vectors carrying puromycin or geneticin resistance markers. Before infection, the cells were seeded in 6-well plates. The negative controls were the corresponding empty lentiviral vectors. After 48-h infection, the cells were selected using puromycin or geneticin, and gene expression was validated using qRT-PCR and western blotting. The siRNAs were synthesized by GenePharma (Shanghai, China) and transfected into cells using Lipofectamine 2000 (Invitrogen, MA, USA). The cells were collected 48 h post-transfection for further experimentation. Tables S1 and S2 list the oligonucleotide sequences.
Protein extraction and western blotting
The cells were lysed in radioimmunoprecipitation assay buffer (Beyotime) and centrifuged at 12,000 rpm for 15 min at 4 °C. The supernatant proteins were quantified using the bicinchoninic acid (BCA) method (Beyotime). An appropriate volume of SDS buffer was added according to the protein concentration, and the samples were heated in a 100 °C bath for 5 min, then cooled on ice for electrophoresis. Proteins were separated using SDS–PAGE and transferred to PVDF membranes. The membranes were blocked with 5% skimmed milk or 1% bovine serum albumin (BSA) and sequentially incubated with primary antibodies and horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology, MA, USA). Protein bands were detected using enhanced chemiluminescence reagents (Advansta, CA, USA) under dark conditions. Table S3 lists the antibodies used.
qRT-PCR
RNA was extracted using TRIzol (Takara, Japan). A PrimeScript RT reagent kit and TB GREEN PriMix (Takara) were used for the reverse transcription and qRT-PCR, respectively. mRNA expression was quantified using a LightCycler 480II system (Roche, Switzerland). The expression data were analyzed using the comparative threshold cycle (2−ΔΔCt) method. Table S4 lists the primer sequences used.
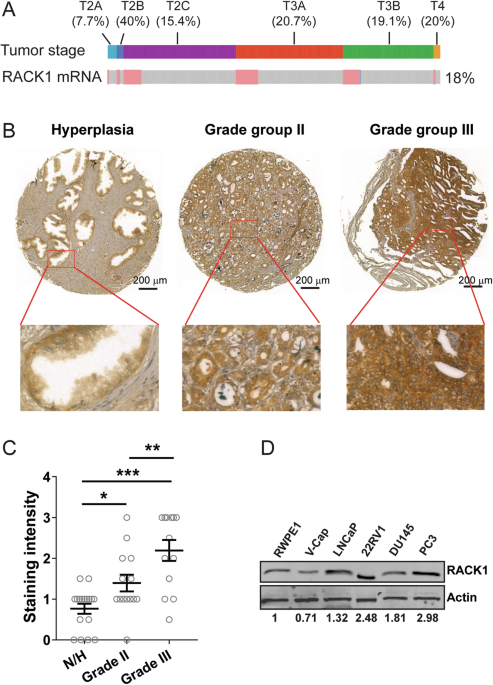
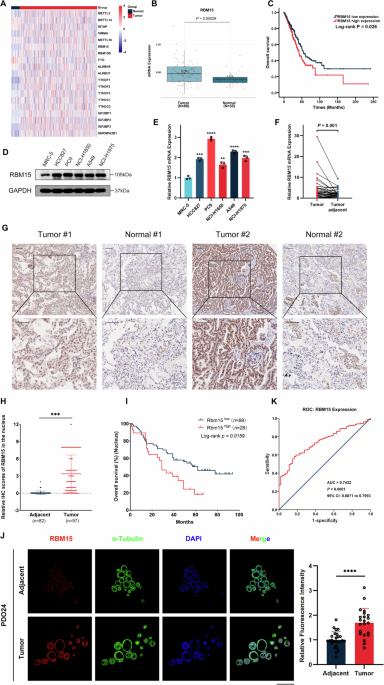
Tissue microarray (TMA), immunohistochemistry (IHC), and TUNEL assay
A LUAD tissue microarray chip containing 98 LUAD tissues and 82 adjacent non-cancerous tissues collected from patients between July 2004 and June 2009 was obtained from Shanghai Outdo Biotech Company (Shanghai, China). The samples were sourced from the National Human Genetic Resources Sharing Service Platform and ethically reviewed by the Shanghai Outdo Biotech Company Ethics Committee (SHYJS-CP-1904014). All experiments were conducted in compliance with relevant guidelines. Informed consent was obtained from all patients.
For the IHC experiments and TMA, animal tissue sections and organoid sections were dewaxed and underwent antigen retrieval, followed by incubation with the appropriate antibodies or TUNEL staining using a cell death detection fluorescein kit (Invitrogen). Imaging was performed using an Aperio scanner for microarrays or an inverted fluorescence microscope (Nikon, Japan). A semi-quantitative evaluation was conducted based on staining intensity and the positive staining percentage. Staining intensity was categorized into 1 (weak positive), 2 (moderate positive), and 3 (strong positive). The percentage of positive cells was classified into 1 (0–10%), 2 (11–50%), 3 (51–80%), or 4 (81–100%). The final score was the product of the staining intensity and percentage of positive cells.
Immunofluorescence
The cells were fixed for 20 min in 4% paraformaldehyde, washed three times with PBS, and permeabilized with 0.2% Triton X-100 for 15 min. After blocking with BSA, the cells were sequentially incubated with primary and secondary antibodies (Invitrogen). Subsequently, the nuclei were stained for 5 min using DAPI. Fluorescent imaging was conducted using a laser scanning confocal microscope (Zeiss, Germany). Table S3 details the antibodies used.
m6A RNA methylation quantification
m6A levels were quantitatively analyzed using an Abcam m6A RNA Methylation Assay Kit (Abcam, ab185912, UK) according to the manufacturer’s instructions. The RNA was bound, capturing m6A RNA, and the signal was detected. Absorbance was measured at 450 nm. The m6A levels were calculated based on the negative and positive controls.
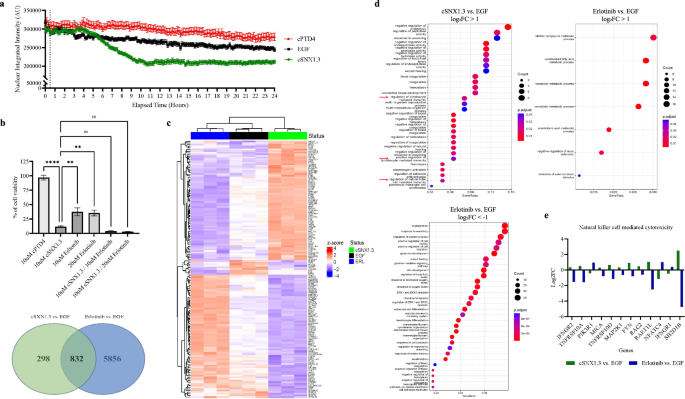
Cell proliferation and drug sensitivity assays
For the cell proliferation assay, 5000 cells per well were seeded in a 96-well plate. A 1:9 dilution of Cell Counting Kit-8 (CCK-8, APExBIO, Houston, USA) solution was added at 0, 24, 48, and 72 h, and the absorbance at 450 nm was measured after incubation at 37 °C. For the drug sensitivity assay, 5000 cells per well were seeded in a 96-well plate and treated for 48 h with osimertinib before a 1:9 diluted CCK-8 solution was added. Absorbance at 450 nm was measured following incubation at 37 °C. The IC50, representing (the osimertinib concentration required to inhibit cell growth by 50%), was calculated using non-linear regression analysis of dose–response data using GraphPad Prism 9.0 (CA, USA).
EdU assay
Cells were seeded at a density of 5000 cells per well in a 96-well plate and, upon reaching 80% confluence, were fixed with 4% paraformaldehyde for 20 min. After three PBS washes, the proliferating cells were labeled using a Click-iT EdU Alexa Fluor 488 Imaging Kit (Invitrogen, C10337). Subsequently, the nuclei were stained for 5 min using DAPI, and the cells were imaged under a fluorescence microscope (EVOS M7000, Invitrogen).
Transwell assay
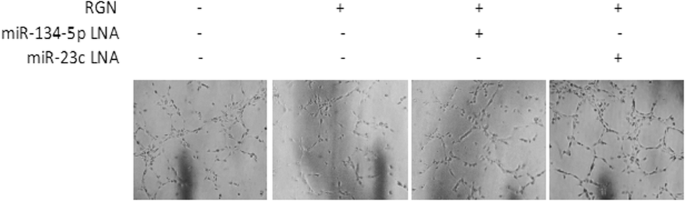
Cell proliferation and migration were assessed by seeding 200 µl single-cell suspension containing 50,000 cells in serum-free medium into the upper Transwell chamber, while 700 µl medium containing 10% FBS was added to the lower chamber. The cells were allowed to migrate for a specified duration in an incubator, then fixed in 4% paraformaldehyde. Following PBS washing, the cells were stained for 15 min with crystal violet. Non-migrating/non-invading cells in the upper chamber were wiped away before imaging under a microscope.
Flow cytometry
Apoptosis was assessed using an Annexin V/fluorescein isothiocyanate (FITC) and propidium iodide (PI) assay kit (Multisciences, Zhejiang, China) according to the manufacturer’s protocol. The apoptosis rates in the cell groups were determined using flow cytometry (BD FACSCanto, NJ, USA). The data were analyzed using FlowJo 10.8.1 (Tree Star, OR, USA).
Methylated RNA immunoprecipitation sequencing (MeRIP-seq)
RNA was extracted, purified, and fragmented into ~100 nucleotide segments using a Magna MeRIP m6A Kit (Millipore, 17-10499, MA, USA). m6A-modified mRNA fragments were enriched using m6A antibody-coated immunomagnetic beads. The complexes were separated, and the enriched RNA was reverse-transcribed into cDNA, which was used for library construction. Sequencing was performed on the Illumina NovaSeq™ 6000 platform with the PE150 sequencing mode. The sequencing services and data analysis were by Hangzhou Kaitai Biotechnology Co., Ltd. (Hangzhou, China).
RNA sequencing (RNA-seq)
The extracted total RNA underwent RNA sample testing, library construction, and library quality control, followed by PE150 sequencing on the Illumina NovaSeq 6000. The sequencing services and data analysis were by Hangzhou Kaitai Biotechnology Co., Ltd.
Immunoprecipitation (IP)
The cells were lysed for 2 h on ice using IP lysis buffer (Beyotime). The protein lysate was centrifuged at 12,000 rpm for 15 min and divided into Input, IP, and IgG aliquots. The protein lysates were incubated with Protein A/G agarose beads pre-bound with antibodies. After washing, the immunoprecipitated protein complexes were separated from the agarose beads, and the RBM15-associated proteins were detected using immunoblots.
RNA immunoprecipitation (RIP)
The RIP assay was conducted using a Magna RIP Kit (17-700, Millipore) according to the kit protocol. Briefly, magnetic beads pre-bound with anti-RBM15 antibody (Proteintech, 66059-1-Ig, IL, USA) or mouse IgG were added to an adequate amount of cell lysate and incubated overnight at 4 °C with rotation. The proteins were digested using protease, and RNA was extracted and purified using TRIzol, followed by reverse transcription into cDNA. The cDNA was analyzed using qRT-PCR.
Methylation inhibitor treatment
The cells were treated for 48 h with 0, 100, or 200 μM 3-deazaadenosine (DAA, APE×BIO, B8470). Subsequently, RNA was harvested and extracted for qRT-PCR analysis.
In vivo tumorigenesis assay
Xenograft models were established using stably transfected H1975 cells and H1975-OR cells with short hairpin RNA negative control (sh-NC), sh-RBM15, and sh-SPOCK1. Male nude mice (4 weeks old) were injected subcutaneously on the right flank with 2 × 106 cells. When the tumors reached a longitudinal diameter of 5 mm, the mice were randomly assigned to each group and subsequently treated orally with an equal volume of osimertinib mesylate (Selleck, S5078, TX, 20 mg/kg/day) or corn oil by gavage every 2 days. The tumor dimensions were measured twice daily. The tumor volume was calculated as follows: (length × width2)/2. The First Affiliated Hospital of Zhejiang University Ethics Committee approved the animal study protocol (2024-1219). All mice were housed in a pathogen-free environment with controlled temperature, maintained on a 12-h light/dark cycle, and had ad libitum access to food and water. Each group comprised at least six animals. Mice that did not develop tumors by 30 days post-subcutaneous injection were excluded from the study. This study does not involve the extent of blinding.
Statistical analysis
All experiments were independently repeated three times. The statistical methods for analyzing database data and clinical samples were determined based on the underlying assumptions and variability. When the variances between the two groups were similar, the Student’s t-test was used; otherwise, the Mann–Whitney test was employed. Data from cell cultures, organoids, and in vivo experiments were analyzed using Student’s t-test. The results are presented as the mean ± SD. Survival analysis was conducted using the Kaplan–Meier method; significance was tested using the log-rank test. All statistical analyses were performed using SPSS 21.0 (IBM, NY, USA). Graphs were created using SPSS 21.0 and GraphPad Prism 9.0. P < 0.05 was considered statistically significant.

留言 (0)