Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. Endoscopic gastric biopsies were obtained from patients at the Nashville Veterans Affairs Medical Center of the VA Tennessee Valley Healthcare System. Patients were undergoing esophagogastroduodenoscopy for clinically indicated reasons and provided informed consent for obtaining research biopsies under VA IRB protocol #1571167 (PI: Keith T. Wilson); clinical data was maintained in REDCap databases. Biopsies were also obtained from patients in Colombia [35]; for this study, individuals provided informed consent for obtaining the biopsies under the Institutional Review Board of Vanderbilt University IRB # 090460 and the Committee on Ethics of Universidad del Valle in Colombia Certificate # 023 of 2002.
The mice were used under protocols V2000018 and V2300022 approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee and the Research and Development Committee of the Veterans Affairs Tennessee Valley Healthcare System. Procedures followed institutional policies, AAALAC guidelines, the AVMA Guidelines on Euthanasia, NIH regulations regarding the Guide for the Care and Use of Laboratory Animals, and the United States Animal Welfare Act of 1996.
Human studies
Biopsies from the gastric antrum and corpus were fixed in 10% neutral buffered formalin, paraffin-embedded, and stained with hematoxylin and eosin (H&E). Each set of biopsies was scored by a gastrointestinal pathologist (MBP) for the presence or absence of chronic gastritis (mononuclear cells) and active gastritis (polymorphonuclear cells), and the microscopic detection of H. pylori. Tissues snap frozen in Brucella broth were subsequently homogenized and cultured for confirmation of the presence of H. pylori as in the mouse studies. Fresh tissues were also used for gastric organoid generation.
Human gastric tissues with low-grade dysplasia (LGD) were derived from adult individuals during a 16-year follow-up from a high cancer risk region in Colombia [35]. The diagnosis of LGD was the consensus reached by at least two out of three GI pathologists in the study.
H. pylori
H. pylori PMSS1, a cagA+ strain with intact type IV secretion system function, was grown on Trypticase soy agar plates containing 10% sheep’s blood [20, 36]. For in vitro and in vivo infections, H. pylori was harvested from the plates and grown overnight in Brucella broth containing 10% fetal bovine serum (FBS). Then, for in vitro studies, the bacteria were resuspended in the media that the cells or organoids were growing in before infection. For infection, the overnight culture of H. pylori was diluted in fresh Brucella broth-FBS, grown, and then collected at the exponential phase.
H. pylori was isolated from endoscopic gastric biopsies from patients at the Nashville Veterans Affairs Medical Center of the VA Tennessee Valley Healthcare System. Briefly, biopsies were manually ground using a pestle in PBS, diluted, plated onto tryptic soy blood agar plates with antibiotics, and grown under anaerobic conditions. Single colonies were isolated and validated through Gram staining. In this study, we used the isolates 18C (cagA+) and 3A (cagA–) that were isolated from the corpus of patient 18 and the antrum from patient 3 in the VA study. Additionally, we utilized PMSS1ΔcagE, an isogenic mutant for the type IV secretion system that fails to translocate the cytotoxin-associated gene A (CagA) [12].
Animals and infections
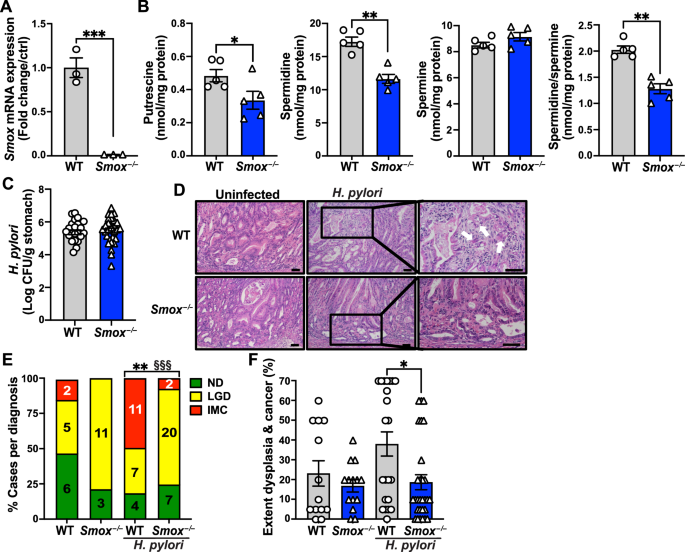
C57BL/6 WT and Smox–/– mice [20, 37] were backcrossed to the FVB/N background and then crossed to transgenic FVB/N insulin-gastrin (INS-GAS) mice. These animals overexpress the human gastrin gene driven by a rat insulin I promoter. Mice were housed in a pathogen-free facility, with ventilated cage racks and were on a 12 h light-dark cycle. Male mice between 8 and 12 weeks were used for all studies. Only male mice were utilized since female mice are protected from H. pylori-induced gastric carcinogenesis [38, 39]. Mice were fed ad libitum with a defined rodent diet AIN-76A (BioServ) one week prior to the first infection and stayed on this diet until the experimental endpoint [21, 22]. Animals were infected by oral gavage with 109 colony forming units (CFU) of H. pylori PMSS1 in 200 μL Brucella broth, two times, on days 0 and 2 [20, 21]. The control mice were gavaged with only broth on both days. In some studies, mice were treated or not with 14 mM spermidine (Sigma) in their drinking water, beginning two days after the second infection and the water was changed weekly [24]. 4 or 8 weeks after the first infection, mice were euthanized and stomachs were harvested as described [36, 40, 41]. Colonization was verified in all infected mice by counting the CFUs cultured after plating serial dilutions of homogenized gastric tissues [20, 36].
We also used gastric tissues from FVB/N INS-GAS animals infected or not with H. pylori PMSS1 ± 3 mg/mL 2-HOBA in the drinking water [22].
Generation of mouse gastric organoids
Stomachs were dissected from WT and Smox–/– animals at the gastroesophageal and gastroduodenal junctions. The forestomach was removed, and the stomachs were butterflied and washed thoroughly with PBS. Stomachs were incubated in 10 mM EDTA at 4 °C for 30 min on a rocker and then transferred to dissociation buffer (43.5 mM sucrose, 54.9 mM D-sorbitol). The tissue was vigorously shaken to release the glands. Gastric glands were plated in Matrigel on pre-warmed 24-well plates and maintained in Advanced DMEM/F12 supplemented with 50% conditioned media from L-WRN murine fibroblast cells [42] with penicillin-streptomycin, gentamicin, 10 μM Y-27632, and 10 μM SB431542. For two-dimensional monolayer culture, organoids were trypsinized and plated on poly-D-lysine and Matrigel coated 8-well chamber slides (Corning) overlaid with 50% conditioned media before infection with H. pylori PMSS1 at a multiplicity of infection (MOI) of 50.
Generation of human gastric organoids
As described [43], organoids derived from adult human stomach biopsies were maintained and passaged. Gastric biopsies were obtained under IRB protocol # 1571167 as described above, and immediately placed in complete DMEM medium on ice in the endoscopy unit until gland isolation in the laboratory. Gastric glands were isolated and gastric epithelial monolayers were generated as described above for the mouse organoids. The epithelial monolayers were infected with H. pylori PMSS1 (MOI 50).
AGS cells
AGS cells were obtained from ATCC, tested for mycoplasma contamination, and maintained in DMEM medium supplemented with 10% FBS, 10 mM HEPES, and 1% penicillin-streptomycin. AGS cells were treated with the SMOX inhibitor MDL 72527 (25 μM) for 24 h before infection, 2-HOBA (100 μM; TSI Co, Ltd.; Lot # SAA20200727) [22] for 2 h prior to stimulation, acrolein (10 μM, NSI Lab Solutions), and/or H. pylori (strains PMSS1, 18C, 3A, or PMSS1ΔcagE; MOI 100).
Histopathology
A gastrointestinal pathologist (MBP) blinded to the experimental groups performed the histologic scoring using the modified Sydney System [44]. For each experimental mouse, a longitudinal strip of stomach tissue, including the corpus and antrum, was fixed in 10% neutral buffered formalin, paraffin-embedded, and stained with H&E. Acute and chronic inflammation in the corpus and antrum of the stomach were determined from 0–3 for each, leading to a final score of 0–12 [20, 36]. The presence and extent of LGD or IMC were determined from H&E sections [21,22,23]. On the mice infected for 8 weeks, the percent loss of parietal cells and extent of mucosa hyperplasia in the corpus on a 0–3 scale [28] was determined from H&E-stained sections.
RNA analysis
RNA was isolated from a longitudinal strip of the mouse stomach, encompassing both the antrum and corpus, using the RNeasy Plus Mini Kit (QIAGEN). RNA was then sequenced as we described [36]. The Ovation RNA Sequencing System V2 (Tecan) was used for the generation and amplification of cDNA. RNASeq library preparation and Next Generation Sequencing (PE150) were performed using the NEBNext Ultra II Directional RNA Library Kit for Illumina (BioLabs, Inc) and Illumina NovaSeq6000 with NovaSeq 6000 SP Reagent Kit (Illumina), respectively. Reads were trimmed to remove the adaptor sequence and read quality was checked via fastq. The transcripts were quantified and mapped to the indexed mouse genome (M23, GRCm38) using Salmon. Transcript-level quantification was then summarized to the gene level, and then annotated and prepared for differential gene expression analysis using the R package tximeta. The R/Bioconductor package DESeq2 was used to identify differentially expressed genes in each group using the Benjamini-Hochberg adjustment (FDR P < 0.05). The complete list of differentially expressed genes (DEGs), including Ensemble transcript identifiers, official gene symbols, fold changes, and P values, is provided in the Supplementary Dataset 1. We used Ingenuity Pathway Analysis software (IPA, QIAGEN) to determine disease functions that were significantly changed with infection, shown in Supplementary Dataset 2. A P value was calculated in IPA using Fisher’s exact test and then converted to a pathway score by converting the P value to the negative log of the P value for each pathway. The predicted activation state and the activation z-score was calculated based on the molecules within each pathway.
We also analyzed RNA expression by RT-real-time PCR. First, cDNA was synthesized using the SuperScript IV Reverse Transcriptase (Thermo Fisher) and Oligo d[T]16 Primer (Thermo Fisher). Then, Power SYBR Green PCR Master Mix (Thermo Fisher) and primers listed in Supplementary Table 1 were used to amplify cDNAs by RT-real-time PCR in the QuantStudio3 (ThermoFischer Scientific).
Polyamine measurement in gastric tissues
Putrescine, spermidine, and spermine were quantified by liquid chromatography-mass spectrometry (LC-MS) using a Thermo TSQ Vantage Triple Quadrupole instrument operated in positive ion mode. As previously described [40], cell pellets were extracted using (70:30) acetonitrile/20 mM NH4OAc pH 8. Extracts were derivatized by reaction with 20 mM dansyl chloride in 100 mM NaHCO3 pH 10 for 20 min. The deuterated internal standards d4-pustrescine, d8-spermidine, and d8-spermine were used to quantify the polyamines [20, 28, 40].
Measurement of acrolein
Acrolein levels were determined in the mouse stomach by LC-MS. Stomach tissues (10–20 mg) were homogenized in 10% acetonitrile (ACN), centrifuged at 8000 rpm for 10 min, and the supernatants were collected. A standard curve was generated by diluting a stock solution of 5 mg/ml acrolein (NSI Lab Solutions) in 90% methanol diluted in ACN. Danzyl hydrazone (10 mg/ml in ACN:10% acetic acid, 2:1; Sigma) was added to the samples and standards (200 μl), vortexed, and then incubated at room temperature for 1 h. The samples were quenched with 100 μl 500 mM glucose in 50% ACN and incubated for 30 min before being transferred into Eppendorf tubes for two extractions with 500 μl ethyl acetate. The top fraction was harvested, dried, and resuspended in 100 μl of 50% ACN. After centrifugation at 2000 rpm for 5 min, the supernatants were analyzed by LC-MS.
Immunostaining
As previously reported [45], paraffin-embedded tissues were sectioned, heated for 1 h at 60 °C, and sections were deparaffinized in xylene and rehydrated in graded alcohols. For immunohistochemistry, endogenous peroxidase was blocked with 3% hydrogen peroxide solution for 30 min at room temperature. Antigen retrieval (Diva Decloaker, BioCare Medial) was performed for 20 min at 110 °C. Tissues were incubated with prediluted rabbit polyclonal anti-Ki-67 [28] (Biocare, Cat # PRM 325 AA), rabbit polyclonal anti-phospho-gamma H2AX [p Ser 139] polyclonal antibody [22] (pH2AX, 1:200, Novus, Cat # NB100-2280), or prediluted rabbit monoclonal anti-myeloperoxidase [28] (MPO, Biocare Cat # PP 023 AA) overnight at 4 °C. The secondary antibody, prediluted HRP Polymer anti-rabbit (DAKO), was applied for 1 h at room temperature. Visualization was performed using 3,3’-diaminobenzidine, and the tissues were counterstained by hematoxylin. The average number of Ki-67-positive and pH2AX-positive epithelial cells per 5 high powered fields in the corpus of the stomach and the average number of MPO-positive cells per 10 high powered fields in the corpus and antrum were quantified by our gastrointestinal pathologist (MBP) in a blinded manner.
For immunofluorescence on mouse and human tissues, paraffin-embedded tissues were sectioned, deparaffinized, and antigen retrieval was performed (DAKO Target Retrieval pH 9). Sections were treated with Proteinase K (DAKO) for 30 seconds, washed with PBS, incubated with 5% normal horse serum (Jackson ImmunoResearch 088-000-001) for 1 h at room temperature, and then incubated with Universal Protein Block (DAKO) for 1 h at room temperature. Tissues were incubated with monoclonal anti-acrolein adduct antibody (1:100, Thermo Fisher, Cat # MA5-27553) overnight at 4 °C. The secondary antibody, donkey anti-mouse IgG, Alexa Fluor 555 (1:600, Thermo Fisher, Cat # A-31570) was applied for 1 h at room temperature and then the slides were mounted with VECTASHIELD HardSet Antifade Mounting Medium with DAPI (Fisher Scientific). Images were taken on a Nikon Eclipse E800 microscope using SPOT 5.4 Software.
We also immunostained AGS cells cultured on chamber slides (LAB-TEK). After treatment or infection, cells were washed with PBS, fixed in 4% paraformaldehyde (ChemCruz) for 20 min at room temperature, washed with PBS, incubated with 100% cold methanol on ice for 5 min, washed with PBS, and blocked with Universal Protein Block (DAKO) for 40 min at room temperature. They were then incubated with the anti-phospho-gamma H2AX [p Ser 139] polyclonal antibody (1:200; Novus, Cat # NB100-2280) overnight at 4 °C followed by an incubation with Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 (1:600; Thermo Fisher, Cat # A32790). Slides were mounted with VECTASHIELD HardSet Antifade Mounting Medium with DAPI (Fisher Scientific). Confocal images were acquired using a Cytation C10 Confocal Imaging Reader and Gen 5+ software (Aligent BioTek). Images were quantified using ImageJ.
AcroleinRED assay
After stimulation with experimental conditions, human and mouse-derived gastric organoids and AGS cells plated on 8-well chamber slides were thoroughly washed with PBS. Each well was incubated with 10 μM AcroleinRED (DiagnoCine, Cat # FNK-FVD-0022) in DMEM for 30 min at 37 °C. All wells were thoroughly washed with PBS, fixed with 3.7% formaldehyde for 30 min at room temperature, washed with PBS, and mounted using the VECTASHIELD HardSet Antifade Mounting Medium with DAPI (Fisher Scientific). Slides were imaged using a Cytation C10 Confocal Imaging Reader and Gen 5+ software (Aligent BioTek). Images were quantified using ImageJ.
Genotyping Helicobacter pylori for cagA
H. pylori was isolated from endoscopic gastric biopsies and grown from a single colony. Bacterial DNA was isolated using the DNeasy Kit for Blood and Tissue kit (Qiagen). The cagA and 16S genes were amplified by RT-real-time PCR using PowerUp SYBR Green Master Mix (Thermo Fisher). Reactions were prepared with 10 ng of DNA and 500 nM primers. Sense and antisense primer sequences and PCR product sizes are as follows: cagA, 5′-GATAACAGGCAAGCTTTTGAGG-3′ and 5′ CTGCAAAAGATTGTTTGGCAGA-3′, 349 bp; 16S, 5′-GGAGTACGGTCGCAAGATTAAA-3′ and 5′-CTAGCGGATTCTCTCAATGTCAA-3′, 127 bp. PCR products were run on a 3% agarose gel with 0.006% SybrSafe DNA Gel Stain (Invitrogen). Stained bands were visualized under UV light and photographed with a digital gel documentation system (Gel Doc EZ Imager and Image Lab Software version 5.1; Bio-Rad Labs Inc.).
Statistics
Prism 10.2.1 (GraphPad Inc.) was used for statistical analysis and all results are expressed as mean ± SEM. At least 3 biological replicates were used for in vitro studies. Data that were not normally distributed according to the D’Agostino & Pearson normality test were log transformed. Outliers were identified using the ROUT test (Q = 5%) and removed from the analysis. Student’s t test was used to determine significant differences between two groups, whereas a one-way ANOVA followed by a Tukey test, or a Kruskal–Wallis and a Mann–Whitney U test was used for multiple groups. A two-way ANOVA followed by a Tukey test was used to determine significant differences between multiple groups on two different variables. Contingency analyses were performed by a Chi-square test or a Fisher’s exact test. For correlation analysis, simple linear regression was used to determine the r and P values.

留言 (0)