
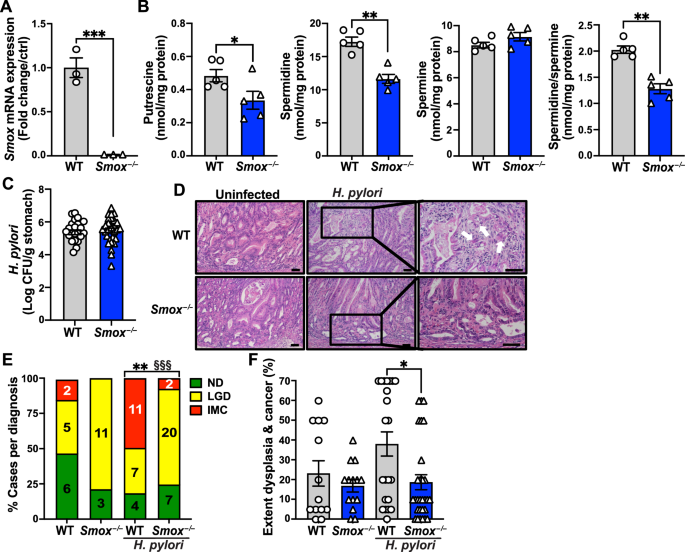
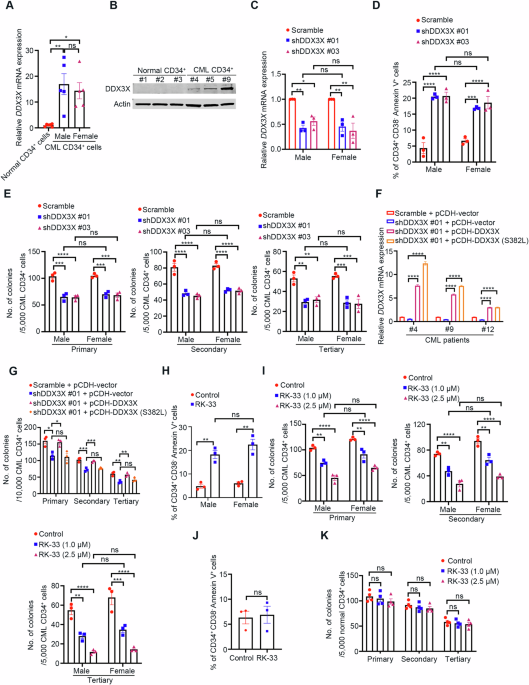
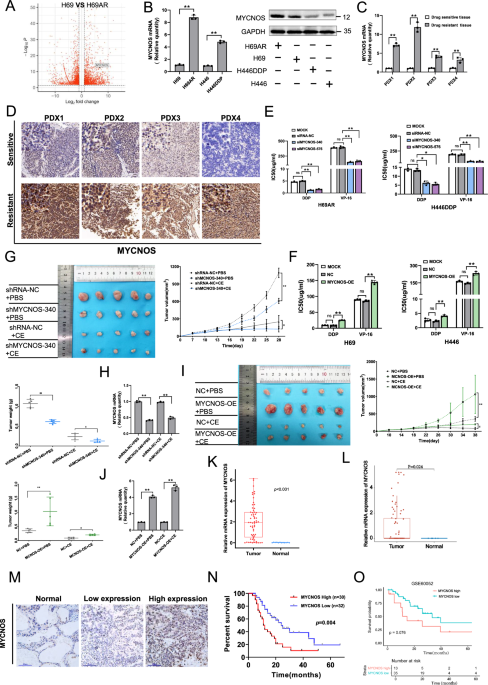
記住我
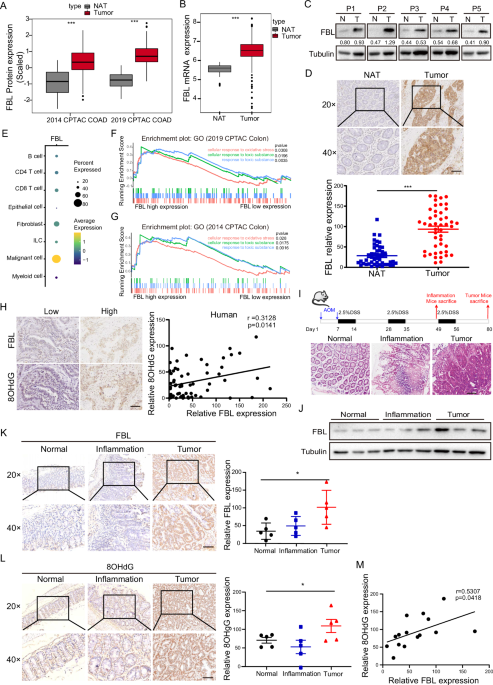
By analyzing two colorectal cancer databases [16, 17], we discovered that FBL was highly expressed in tumor tissues compared to normal tissues at both the proteomic and mRNA levels (Fig. 1A, B). To verify FBL expression in human CRC, we collected tumor tissues and paired normal tissues adjacent to tumors (NATs) from 46 patients. Consistent with the analysis, FBL expression was elevated in tumor samples compared to NATs, as confirmed by both immunoblotting (Fig. 1C) and immunohistochemical (IHC) staining (Fig. 1D). In addition, we observed that FBL expression was predominantly enriched in tumor sites rather than stromal sites. The scRNA-seq analyses for colorectal cancer further validated that FBL was highly enriched in malignant cells compared to other cell clusters, highlighting the significant roles of FBL in tumor cells [18] (Fig. 1E).
Fig. 1: FBL is highly expressed in colorectal cancer and responds to oxidative damage.
A The protein expression of FBL in tumors and NATs analysed in 2019 and 2014 CPTAC COAD databases. NATs, non-cancerous adjacent tissue. (***p < 0.001, Wilcoxon rank-sum test). B The mRNA level of FBL in tumors and NATs analysed in TCGA-COAD project. (***p < 0.001, Wilcoxon rank-sum test). C Immunoblotting analysis of FBL protein level in T (tumor) samples and paired N (non-cancerous adjacent tissue) from CRC patients. D Immunohistochemical stains of FBL in tumor samples and paired NATs from CRC patients. Representative images of NAT and tumor sample (up) and Quantification of FBL expression (down) were shown. (*p < 0.05, **p < 0.01, ***p < 0.001, error bar = ±SEM, n = 46, scale bar =100 μm). E Bubble heatmap showing the expression of FBL in each Smart-seq2 cell type of CRC single cell databases (GSE146771). Dot size indicates the percentage of expressed cells, colored by average expression levels. F GSEA pathway analysis of proteins enriched in patients with FBL high expression and in patients with FBL low expression in 2019 CPTAC COAD databases. G GSEA pathway analysis of proteins enriched in patients with FBL high expression and in patients with FBL low expression in 2014 CPTAC COAD databases. H Representative image (left) and Pearson correlation analysis (right) of immunohistochemical stains of FBL and 8-OHdG in 61 CRC patients’ tumors. (*p < 0.05, error bar = ±SEM, scale bar =100 μm). I Schematic diagram of colitis-associated carcinoma C57 mouse model induced by AOM/DSS. The HE staining of normal, inflammatory and tumor intestinal tissues in mice sacrificed as indicated after induction. J Immunoblotting analysis of FBL protein levels in normal, inflammatory and tumor intestinal tissues of mice. K Representative images (left) and quantification (right) of immunohistochemical stains of FBL in normal, inflammation and tumor intestinal tissues of mice. (*p < 0.05, error bar = ±SEM, n = 5, scale bar =100 μm). L Representative images (left) and quantification (right) of immunohistochemical stains of 8-OHdG in normal, inflammation and tumor intestinal tissues of mice. (*p < 0.05, error bar = ±SEM, n = 5, scale bar =100 μm). M Pearson correlation of FBL and 8-OHdG staining in normal, inflammation and tumor intestinal tissues of mice.
To further investigate the functional involvement of FBL in colorectal tumors, we analyzed FBL-involved pathway enrichments in three proteomic colorectal cancer databases [16, 17, 19]. Interestingly, we observed, for the first time, that the expression of FBL positively correlated with toxic substrates and oxidative stress (Fig. 1F, G, Fig. S1A). Oxidative stresses are known to attack genomic guanine, leading to the formation of 8-hydroxy-2’-deoxyguanosine (8-OHdG) or 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxodG), thereby causing oxidative DNA damage. To explore the possible correlations of FBL with oxidative DNA damage, we stained 8-OHdG and FBL in patient tumor samples. Our results demonstrated a positive correlation between FBL expression and 8-OHdG levels (Fig. 1H). To further investigate the association of FBL with oxidative DNA damage and tumorigenesis, we established an azoxymethane/dextran sulfate sodium (AOM/DSS) colitis-associated carcinoma murine model, which recapitulates inflammatory colorectal cancer, and validated pathogenic tissues through HE staining (Fig. 1I). We detected the protein level of FBL in normal, inflammatory, and tumor intestinal tissues and found that FBL expression gradually increased concomitantly with the accumulation of toxic agents, ultimately exhibiting the highest expression in tumors, as demonstrated by both immunoblotting and IHC staining (Fig. 1J, K). Furthermore, we stained for 8-hydroxyguanosine (8-OHdG) in normal, inflammatory, and tumor tissues from the AOM/DSS mouse model. We observed that the level of 8-OHdG increased during tumor development and that FBL expression positively correlated with 8-OHdG staining (Fig. 1L, M). Thus, our data provide evidence that the expression of FBL increases concomitantly with oxidative stresses and is associated with oxidative DNA damage.
FBL acts as a critical factor in oxidative DNA damage repair and is directly recruited to DNA damage sitesTo elucidate the impact of FBL on oxidative damage, we treated FBL-overexpressing colorectal cancer cells with oxidative stresses and observed that cells with FBL overexpression were more resistant to hydrogen peroxide (H2O2) or KBrO3-induced oxidative damage (Fig. 2A, B, Fig. S2A). In contrast, either RKO or SW480 cells with FBL knockdown were hypersensitive to oxidative damage (Fig. 2C, D, Fig. S2B–E). To further investigate the direct impact of FBL on oxidative stress-induced DNA damage, we performed alkaline comet assays, where the tail length reflects the level of DNA breaks. The results revealed reduced DNA breaks in FBL-overexpressing cells (Fig. 2E) and, conversely, exacerbated breaks in FBL knockdown cells under H2O2 treatment (Fig. 2F).
Fig. 2: FBL acts as a critical factor in oxidative DNA damage repair and is directly recruited to DNA damage sites.
Cell viability of HCT8 cell lines stably overexpressing Flag or Flag-FBL. The cells were exposed to H2O2A or KBrO3B at concentrations as indicated for 48 h and then analysed by CCK8 assay. (*p < 0.05, ***p < 0.001, error bar = ±SEM, n = 5). Cell viability of RKO cell lines stably expressing control shRNA (shctrl) or FBL-targeted shRNA (shFBL). The cells were exposed to H2O2C or KBrO3D at concentrations as indicated for 48 h and then analysed by CCK8 assay. (***p < 0.001, error bar = ±SEM, n = 5). E Alkaline comet assays for HCT8 cell lines stably overexpressing Flag and Flag-FBL. Cells were treated with 1 mM H2O2 for 20 min and then harvested 1 h after recovery. Representative images (left) and quantification (right) were shown. (***p < 0.001, error bar = ±SEM, n = 20–50, scale bar =20 μm). F Alkaline comet assays for RKO cell lines stably expressing shctrl, shFBL #1 or shFBL#2. Representative images (left) and quantification (right) were shown. (***p < 0.001, error bar = ±SEM, n = 20–50, scale bar =20 μm). G The recruitments of EGFP-FBL to the DNA damage sites induced by 405 nm microirradiation in U2OS cells. Yellow arrows represent laser points/lines induced by 405 nm laser. (scale bar =2.5 μm). H Schematic representation of the FBL truncations (left). FL, FBL Full-length; ΔGAR, truncated mutants lacked residues 8–80 aminol acid (aa) for glycine and arginine rich (GAR) domian; ΔRB, mutant lacked residues 133–222 aa for RNA binding (RB) domain; Δα-helical, mutant lacked residues 274-306 aa. I The recruitments of EGFP-FBL-FL, EGFP-FBL ΔGAR, EGFP-FBL ΔRB, EGFP-FBL Δα-helix to the DNA damage sites induced by 405 nm microirradiation in U2OS cells. (scale bar =2.5 μm,). J The quantification of time-lapse recruitments of EGFP-FBL FL and truncations to DNA damage sites induced by 405 nm microirradiation in U2OS (right). (***p < 0.001, error bar =±SEM, n = 10–15 cells).
To decipher the role of FBL in DNA damage repair, we conducted laser microirradiation to generate localized DNA damage in U2OS cells and then visualized FBL accumulation. Live-cell imaging demonstrated that EGFP-tagged FBL was recruited to the sites of laser-induced DNA damage immediately after microirradiation in the nucleoplasm (Fig. 2G). Analyzing the FBL structure, we observed two main domains: the N-terminal domain and the methyltransferase domain. The N-terminal domain, known as the GAR domain, was defined as a disordered region enriched in arginine and lysine. The methyltransferase domain consisted of the RNA-binding (RB) domain and the α-helix domain (Fig. 2H). To determine which domain is needed for the DNA damage response of FBL, we then constructed three truncations: FBL truncated GAR (ΔGAR), RB (ΔRB) or α-helix domain (Δα-helix). Intriguingly, the damage response of the different truncations showed that GAR was a domain required for DNA damage recruitment of FBL, as evidenced by the fact that FBL-truncated GAR completely lost laser microirradiation-induced accumulation. The other truncations of FBL maintained the DNA damage response (Fig. 2I, J).
PARP1 serves as the upstream regulator of the DNA damage response of FBLThe initiation of DNA damage repair and pathway choice relies on the mediation of a series of early and upstream regulators, including members of the phosphatidylinositol 3 kinase (PI3K)-like kinases (PIKK) family, such as ataxia-telangiectasia mutated (ATM), Rad3-related protein (ATR), DNA-dependent protein kinase (DNA-PK), and DNA damage sensor poly-(ADP-ribose) polymerase 1 (PARP1). To explore the upstream factors that might regulate FBL-related DNA damage repair, we pretreated the cells with ATM, ATR, DNA-PK or PARP inhibitors individually before laser microirradiation. Interestingly, the DNA damage response of FBL was completely abolished by a PARP inhibitor (olaparib) and partially inhibited by ATR inhibitors but remained unchanged under treatment with other inhibitors in both U2OS and RKO colon cancer cell (Fig. 3A, Fig. S3A). Olaparib targets both PARP1 and PARP2. To further characterize which PARP family members are necessary for the DNA damage response of FBL, we investigated the recruitment of FBL after laser microirradiation in PARP1 or PARP2 knockdown cells or knockout cells. The results suggested that depletion of PARP1, but not PARP2, significantly reduced the accumulation of FBL at DNA damage sites (Fig. 3B–D, Fig. S3B). Meanwhile, we demonstrated that endogenous FBL accumulated at damage sites following microirradiation-induced damage. This accumulation was further prevented by either a PARP inhibitor or PARP1 knockdown (Fig. S3C). In addition, recruitment of FBL was recovered in PARP1 -/- cells rescued by the PARP1 WT, but not PARP1 kinase dead mutants, E988A or W318R (Fig. 3E). Furthermore, olaparib or PARP1 knockdown did not affect the expression of FBL (Fig. S3D). These data collectively suggest that PARP1 serves as the upstream regulator of the DNA damage response of FBL.
Fig. 3: PARP1 serves as the upstream regulator of the DNA damage response of FBL.
A The representative images (left) and quantification of normalized fluorescent intensity (right) of EGFP-FBL recruited to DNA damage sites induced by 405 nm microirradiation in U2OS cells pretreated with DMSO (control) or inhibitors as indicated. ATMi: ATM inhibitor KU55933 (10 μM for 1 h), ATRi: ATR inhibitor AZD6738 (1 μM for 24 h); DNA-PKi: DNA-PK inhibitor PI103 (2.5 μM for 24 h); PARPi: PARP inhibitor Olaparib (5 μM for 24 h). (***p < 0.001, error bar =±SEM, n = 10–15 cells, scale bar =2.5 μm). B The recruitments of EGFP-FBL to the DNA damage sites induced by microirradiated in U2OS transfected with siRNA as indicated (left). Intensity dynamics of EGFP-FBL recruited to DNA damage sites induced by 405 nm microirradiation were shown (right, ***p < 0.001, error bar =±SEM, n = 10–15 cells, scale bar =2.5 μm). C The mRNA expression levels of PARP1 and PARP2 in cells transfected with non-targeting siRNA (sictrl), siRNA targeting at PARP1 (siPARP1) and PARP2 (siPARP2) analysed by qPCR were shown (***p < 0.001, error bar = ±SEM, n = 3). D The representative images (top) and the intensity dynamic analysis (bottom) of EGFP-FBL recruited to the DNA damage sites induced by 405 nm microirradiation in HeLa cells (WT) or PARP1 knockout (KO) HeLa cells. (***p < 0.001, error bar = ±SEM, n = 10–15 cells, scale bar =2.5 μm). E The representative images of EGFP-FBL recruited to the DNA damage sites induced by 405 nm microirradiation in PARP1 KO HeLa cells co-transfected EGFP-FBL with RFP-empty, RFP-PARP1, RFP-PARP1E988A or RFP-PARP1W318R respectively. (scale bar =2.5 μm).
FBL could either be PARylated by PARP1 or recognize PARylated proteinsTo determine how PARP1 regulates FBL, we first employed a bimolecular fluorescence complementation (BiFC) assay to detect the interaction of PARP1 and FBL in live cells [20]. PARP1 and FBL were fused with the N-terminal (YFPn, residues 1–155) and C-terminal (YFPc, residues 156–239) fragments of the Venus variant of yellow fluorescent protein (YFP), respectively (Fig. 4A). A strong yellow fluorescent signal was observed at laser-induced DNA damage sites in both U2OS and RKO CRC cells transfected with PARP1-YFPc and FBL-YFPn, confirming their interaction. However, no fluorescence was observed in cells with the fusion protein alone (Fig. 4A, Fig. S4A). Furthermore, we confirmed the interactions between FBL and PARP1 through reciprocal coimmunoprecipitation (Fig. 4B, C). In addition, H2O2 treatment greatly enhanced the interaction of FBL and PARP1 (Fig. 4D). We previously demonstrated that the GAR domain is crucial for the DNA damage response of FBL. We next investigated whether the interaction of FBL and PARP1 relies on the GAR domain. Using an in vitro pull-down assay, we purified GST-tagged FBL (GST-FBL) or GST-tagged FBL truncated GAR (GST-ΔGAR) proteins from bacteria and incubated them with the PARP1 protein individually. The results showed that only GST-FBL interacted with and pulled down PARP1, while GST-ΔGAR failed to interact (Fig. 4E), indicating that FBL interacts with PARP1 directly through the GAR domain.
Fig. 4: FBL interacts with PARP1 through the GAR domain and is PARylated by PARP1.
A Schematic illustration of bimolecular fluorescence complementation (BiFC) assay (left). FBL was fused to the N-terminal of YFP (YFPn) (FBL-cYFPn). PARP1 was fused to the C-terminal of YFP (YFPc) (PARP1-cYFPc). The images of YFP signal at DNA damage sites induced by 405 nm microirradiation in cells co-transfected FBL-cYFPn with YFPn or PARP-cYFPc, or PARP-cYFPc with YFPn (right) in U2OS cells. B Co-immunoprecipitation of Flag-SBP-FBL with endogenous PARP1 in HEK293T cells. C Co-immunoprecipitation of Flag-SBP-PARP1 with endogenous FBL in HEK293T cells. D Co-immunoprecipitation of Flag-SBP-PARP1 with endogenous FBL in HEK293T cells with or without 1 mM H2O2 treatment for 10 min or 20 min as indicated. E In vitro binding assay for GST-FBL with PARP1. Bacterial purified GST, GST-FBL and GST-FBL ΔGAR proteins were incubated with PARP1 protein overnight and then precipitated with GST sepharose beads. F In vitro binding assay for GST-FBL with PAR polymer. Bacterial purified GST, GST-FBL and GST-FBL ΔGAR proteins were incubated with PAR polymer overnight and then precipitated with GST sepharose beads. G In vitro PARylation assay for GST-FBL by PARP1. H The recruitments of EGFP-FBL-FL, EGFP-FBLΔ21-27 aa (amino acids), EGFP-FBL Δ28-40 aa, EGFP-FBL Δ46-59 aa, EGFP-FBLΔ70-78 aa to the DNA damage sites induced by 405 nm microirradiation in U2OS cells(left). (scale bar =2.5 μm). The quantification of time-lapse recruitments of EGFP-FBL FL and truncations to DNA damage sites induced by 405 nm microirradiation in U2OS (right). (***p < 0.001, error bar =±SEM, n = 10–15 cells). I The schematic illustration for constructing Flag-SBP-Af1521 (Macro domain) (top). The immunoblotting analysis of FBL were preformed in Flag-SBP-Af1521 precipitants in HEK293T cells with or without 2 mM H2O2 treatment for 20 min (bottom). The PJ34 (40 mM) was preincubated for 1.5 h before H2O2 treatment. J The schematic illustration of FBL PARylated by PARP1 or recognizing PARylated protein.
PARP1 is activated by DNA lesions and plays a crucial role as an early sensor for DNA damage. Activated PARP1 subsequently PARylates numerous DNA damage response (DDR)-related proteins, amplifying repair cascades [21,22,23]. The PARP1-dependent DNA damage response of FBL suggests two possibilities: FBL may act as a PARylated substrate catalyzed by PARP1, or FBL may recognize PARylated proteins. To explore these possibilities, we first examined the in vitro PARylation of FBL. We observed that FBL was PARylated by PARP1 only under the condition of complete supplementation of DNA and NAD+ (Fig. 4F), while FBL showed no apparent PARylation under incubation with either DNA or NAD+ alone. Consistent with in vitro assay, we observed increased PARylation of pull-down FBL after PARP1 overexpression in response to H2O2 treatment in the SW480 CRC cell line (Fig. S4B). We then investigated the interaction of FBL with PARylation and found that FBL directly bound to PAR through its GAR domain in vitro (Fig. 4G). To demonstrate whether the recognition of PAR by FBL or PARylated FBL occurs in vivo, we employed two PAR binding macro domains, PBZ (PAR-binding zinc finger) and the AF1521 macrodomain, which recognize and bind PAR with high affinity [24,25,26]. We fused PBZ and FBL with YFPc and YFPn (PBZ-YFPc and FBL-YFPn) and coexpressed them in U2OS cells, observing bright fluorescent signals accumulated at laser-induced DNA damage sites, similar to the observations in Fig. 4A (Fig. S4C). In addition, we fused PBZ with mRuby fluorescent protein (PBZ-mRuby) and coexpressed PBZ-mRuby with PARP1-YFPc and FBL-YFPn. As expected, under laser microirradiation-induced DNA damage, PBZ-mRuby accumulated at the DNA damage site and colocalized with the FBL/PARP1 YFP signal (Fig. S4D). Next, we used another PAR-recognizing domain, Af1521, and performed an Af1521-directed pull-down experiment to obtain extensive PARylated proteins [6]. FBL was detected in Af1521 precipitates (Fig. 4I). Furthermore, the FBL signal was enhanced following H2O2 treatment and completely abolished under pretreatment with the PARP inhibitor PJ34 (Fig. 4I).
To further explore the PARylation sites, we mutated specific amino acids including Asp, His, Ser, Arg in the GAR domain according to preferential ADP-ribosylation sites for PARP1 [7]. However, none of these mutants completely abolished the DNA damage response of FBL (Fig. S4E, F). We then sequentially deleted several amino acids in the GAR domain and validated the DNA damage response of all mutants. Four mutants with deletions at amino acids Δ21-27, Δ28-40, Δ46-59, and Δ70-78 exhibited attenuated DNA damage response (Fig. 4H, Fig. S4G), indicating that PARP1 regulates the damage response of FBL through the entire GAR domain of FBL or multiple sites. Taken together, these results indicate that FBL could either be PARylated by PARP1 or recognize PARylated proteins (Fig. 4J).
FBL facilitates short-patched base excision repair pathwayAssuming that oxidative damage repair and subsequent base excision repair (BER) pathways play key roles in repairing oxidative damage, we carried out a BER assay [27] and observed that the repair efficiency within BER pathways was diminished in FBL-knockdown (shFBL) cells compared with control (shctrl) cells (Fig. 5A). However, this reduction was reversed upon reintroduction of FBL into the cells (Fig. 5B).
Fig. 5: FBL facilitates short-patched base excision repair pathway.
A The schematic illustration of BER efficiency assay (left). The BER efficiency assay (the ratio of GFP+ cells to DsRed+ cells analysed by FACS after 48 h transfection) in shctrl or shFBL (#1, #2) RKO cell lines (right) (**p < 0.01, error bar = ±SEM, n = 3). B The BER assay in shctrl, shFBL RKO cells stably expressing Flag empty vectors or shFBL RKO cells rescued by shFBL resistant Flag-FBLR. (*p < 0.05, **p < 0.01, error bar = ±SEM, n = 3). The recruitments (left) and intensity analyses (right) of EGFP-Lig III C EGFP-FEN1 D and EGFP-PCNA E recruited to the DNA damage sites induced by 405 nm microirradiation in shFBL#2 RKO cells stably expressing Flag-empty, Flag-FBLR or Flag-FBLΔGAR R, respectively. (*p < 0.05, **p < 0.01, error bar = ±SEM, n = 10–15 cells, scale bar = 2.5 μm).
We then examined the response of key factors involved in processing oxidative DNA damage and the BER pathway to determine the downstream effects of FBL regulation. The results showed that DNA Ligase III (Lig III), a factor in the short-patch BER pathway, accumulated rapidly in cells supplemented with FBL. Conversely, the damage response of factors involved in the long-patch BER pathway, such as flap structure-specific endonuclease 1 (FEN1), proliferating cell nuclear antigen (PCNA), and Replication Factor C Subunit 1 (RFC1), was quicker to disassociate in FBL-supplemented cells compared to FBL-knockdown cells (Fig. 5C–E, Fig. S5A, B). However, factors associated with the short-patch BER pathway, like X-ray repair cross complement 1 (XRCC1) and polymerase β (Polβ), did not exhibit altered DNA damage response (Fig. S5C, D).
Furthermore, we observed rapid disassociation of 8-oxoguanine DNA glycosylase 1 (OGG1) in cells supplemented with FBL, while OGG1 was retained at damage sites in FBL-deficient cells or cells rescued only by FBL GAR truncation (Fig. S5E). The response of apurinic/apyrimidinic endodeoxyribonuclease 1 (APEX1) and Nth-like DNA glycosylase 1 (NTHL1) did not show significant changes between FBL-knockdown cells and FBL-rescued cells (Fig. S5F–G). Additionally, we observed a negative correlation between FBL expression and single-nucleotide polymorphisms (SNPs) in the TCGA (The Cancer Genome Atlas Program) CRC database (Fig. S5H). These findings collectively suggest that FBL facilitates short-patch BER while suppressing long-patch BER.
Elevated expression of FBL reduces the accumulation of DNA breaks during oxidative stressThe balance between DNA damage and repair capacity is critical for maintaining genome integrity, as excessive oxidative damage beyond repair capacity can result in the formation of double-strand DNA breaks. Phosphorylation of the histone variant H2AX at site Ser 140, known as γH2AX, serves as an early indicator of double-strand DNA damage [28]. Utilizing laser microirradiation-induced DNA damage, we found an intriguing negative correlation between the intensity of exogenous or endogenous FBL and γH2AX in U2OS and HCT8 cell lines (Fig. 6A, Fig. S6A). To evaluate the repair dynamic, we then examined the kinetics of γH2AX in cells with either overexpressed or depleted levels of FBL. Remarkably, FBL overexpression led to the rapid disappearance of γH2AX foci following oxidative damage (Fig. 6B). A similar result was obtained by γH2AX western blotting. (Fig. 6C). Conversely, in FBL-deficient cells, γH2AX levels remained sustained and gradually declined (Fig. S6B, C), a trend that was reversed upon FBL complementation (Fig. 6D). To validate the relationship between FBL and γH2AX in carcinogenesis, we examined FBL and γH2AX expression in inflammatory and tumor samples from the AOM/DSS colitis-associated carcinoma murine model. Intriguingly, we observed a negative correlation between FBL expression and γH2AX levels during tumorigenesis (Fig. 6E, F), suggesting that FBL plays an indispensable role in preventing DNA double strand breaks.
Fig. 6: High expression of FBL reduces the level of DNA breaks under oxidative stress.
A Representative images (left) and Pearson correlation (right) of the fluorescence intensity of EGFP-FBL (green) merged with immunofluorescent stain of γH2AX (red) in U2OS cells immediately after microirradiation. (scale bar =2.5 μm). B The percentage of HCT8 cell lines stably overexpressing Flag and Flag-FBL with positive γH2AX foci. The cells were treated with 1 mM H2O2 for 20 min and then recovery and collected at indicated time points. Cells with ≥5 foci were defined as positive cells. The fold changes were calculated by normalized the time point collected to the time points immediately after drug withdrawal (0 h). (***p < 0.001, error bar = ±SEM, scale bar =10 μm, n ≥ 300 cells). C Immunoblotting analysis of γH2AX in HCT8 cell lines stably expressing Flag or Flag-FBL. D Immunoblotting analysis of γH2AX in shctrl, shFBL knockdown RKO cell lines or shFBL knockdown cell lines rescued by Flag-FBLR. E, F Representative image E and Pearson correlation F of immunohistochemical stains of paired γH2AX and FBL in inflammation and tumor intestinal tissues of mice. (scale bar =100 μm). G, H Alkaline comet assays for HCT8 cell lines stably overexpressing Flag and Flag-FBL. Cells were treated with 1 mM H2O2 for 20 min and then harvested 1 h after recovery or 50uM olaparib 48 h as indicated. Representative images G and quantification H were shown. (***p < 0.001, error bar = ±SEM, n > =30, scale bar =20 μm).
The discovery of PARP as an upstream regulator of FBL in the DNA damage response opens new possibilities for application of PARP inhibitor (PARPi) in CRC with high FBL expression. PARP inhibitors (PARPis) have been extensively used in cancers with HRD [5], while their potential in colorectal cancers is still under investigation. We assessed the extent of DNA breaks in cells overexpressing FBL under oxidative stress alone or in combination with PARP inhibition. Our results showed a reduction in DNA breaks in FBL-overexpressing cells treated with H2O2 alone, but an exacerbation of DNA breaks was observed under treatment with both H2O2 and PARPi (Fig. 6G, H). These findings indicate that high expression of FBL mitigates the extent of DNA breaks induced by oxidative stress. Moreover, inhibiting FBL function through PARP inhibition could exacerbate the DNA damage caused by oxidative stress.
PARP inhibitor enhances radiotherapy in FBL highly expressed colorectal cancerLow doses of ionizing radiation (IR) induce cell death through accumulated oxidative damage. We evaluated the impact of treating cancer cells and tumors overexpressing FBL with a PARPi alone or in combination with a low dose of ionizing radiation (IR). The results showed that treatment with IR or olaparib alone modestly reduced the survival of cells expressing either empty vectors or FBL (Fig. 7A, B). However, combining PARPi with IR significantly decreased the viability of FBL-overexpressing cells compared to control cells (Fig. 7A, B). To further investigate this therapeutic approach in tumors, we established a xenograft mouse model using FBL-overexpressing cells or vector cells (Fig. 7C). Treatment of tumor-bearing mice with IR, olaparib, or the combination of IR with olaparib resulted in slightly reduced tumor growth. Notably, FBL-overexpressing tumors exhibited increased sensitivity to the combination of IR with olaparib compared to the vector group (Fig. 7D–F, Fig. S7A). In addition, immunohistochemical staining of γH2AX in residual tumors indicated a significant increase in DNA damage in FBL high-expressing tumors compared to other groups following combined treatment (Fig. 7G, H, Fig. S7B), highlighting the potential alternative application of PARPi in combination with radiotherapy in FBL high-expressing CRC.
Fig. 7: PARP inhibitor enhances radiotherapy in FBL highly expressed colorectal cancer.
A Cell viability of HCT8 cell lines stably overexpressing empty vector Flag or Flag-FBL. The cells were exposed to 10 μM olaparib or 2 Gy IR as indicated and then analysed by CCK8 assay. (*p < 0.05, ***p < 0.001, error bar = ±SEM, n = 6). B Cell viability of SW480 cell lines stably overexpressing empty vector Flag or Flag-FBL. The cells were exposed to 10 μM olaparib or 2 Gy IR as indicated and then analysed by CCK8 assay. (*p < 0.05, ***p < 0.001, error bar = ±SEM, n = 6). C Schematic diagram of nude mice implant model and treatment schedule. Mice of HCT8 Flag/ Flag-FBL cell line were randomized to 4 groups as indicated (n = 5): (1) the vehicle group (10%DMSO / 30%PEG300 / saline solution for 3 days by intraperitoneal injection); (2) the olaparib group (olaparib (50 mg/kg/day) for 3 days by intraperitoneal injection); (3) the irradiation group (10%DMSO / 30%PEG300 / saline solution for 3 days by intraperitoneal injection, tumor-localized irradiation (2 Gy) given 1 h after the dose of DMSO); (4) the combination group (olaparib (50 mg/kg/day) for 3 days by intraperitoneal injection, tumor lacalized irradiation (2 Gy) given 1 h after the dose of olaparib). D Tumors isolated from individual mice. E Growth curve of xenograft tumors derived from indicated group of Flag and Flag-FBL HCT8 cells. Tumor volumes were measured at indicate time points. (n = 5, Error bars represent SEM, *p < 0.05, **p < 0.01, ***p < 0.001, two-sided, unpaired t-test). F The tumor weight measured at the end of the treatments (n = 5, Error bars represent SEM, *p < 0.05, *** P < 0.001, two-sided, unpaired t-test). G Representative image of immunohistochemical (IHC) staining for DNA damage maker γH2AX in tumor samples derived from indicated xenografts in nude mice. Scale bar, 100 µm. H Quantitation of the normalized histological score (H score) of γH2AX with three individual view for each tumor in different treatment groups as indicated.
留言 (0)