
記住我
In colorectal cancer, increased protein stability of the WNT effector β-Catenin is critical for tumorigenesis and mediated either by loss of function mutations and truncations within the APC gene or via mutations of the degron motive within CTNNB1, the gene encoding β-Catenin. While dysregulation or mutation of upstream canonical regulators of β-Catenin result in aberrant WNT target gene expression patterns due to increasing the protein levels β-Catenin [23, 30], did we wonder if the ubiquitylation of the WNT effector itself is altered [16], as several additional ubiquitin-regulatory pathways have been reported in the past that could contribute to oncogenesis in CRC.
To investigate whether the nature of CTNNB1 or APC mutations impact the stability and ubiquitylation of β-Catenin in CRC, did we conduct an endogenous ubiquitin TUBE (tandem ubiquitin binding entity) assay in a panel of human CRC lines, comprising β-Catenin mutant lines (HCT116 and LS174T), or cell lines varying in the truncation length of APC, DLD-1, SW480, SW620, Colo320 and HT-29, respectively (Fig. 1a). Remarkably, and irrespective of genetic alteration we were able to detect poly-ubiquitylation of β-Catenin (Fig. 1a). This suggested that protein stability of the WNT effector could be altered in a Ubiquitin Proteasome System (UPS) specific fashion likely by additional factors. To discover such regulators, we performed a human DUB siRNA screen in the APC truncated CRC line HT-29, followed by assessing the residual protein abundance of endogenous β-Catenin by immunofluorescence imaging using a high content screening microscope setup, relative to siRNA control transfected HT-29 (Fig. 1b, c). Analysis of the screen not only identified known regulators of β-Catenin stability, such as USP20 or UCHL1, but highlighted the deubiquitylase USP10 as a positive regulator of β-Catenin stability, along with previously identified DUBs (Fig. 1c and Supplementary Fig. S1a, b). Indeed, we verified that loss of USP10 resulted in the depletion of the cytosolic and nuclear pool of β-Catenin, as seen by immunofluorescence (Fig. 1c).
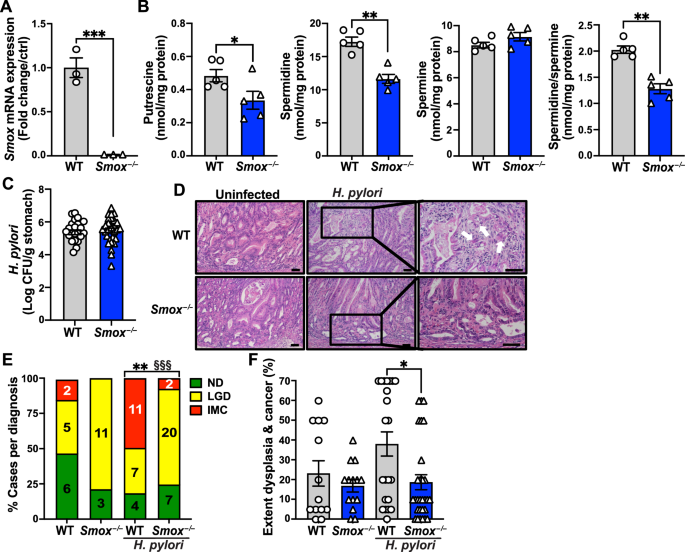
Fig. 1: USP10 is a novel regulator of β-Catenin signalling in CRC and correlates with poor patient survival.
a Tandem Ubiquitin Binding Entity (TUBE) assay of endogenous poly-ubiquitylated proteins, followed by immunoblotting against endogenous β-Catenin in human CRC cell lines with varying mutations. HCT116 and LS174T mutant for β-Catenin, DLD-1, SW480, SW620, Colo320 and HT-29 mutant for APC. β-Actin served as loading control. b Schematic model of siRNA DUB library screen conducted in APC mutant HT-29 cells. Cells were transfected with 4 individual siRNA against DUBs and 48 h post transfection immunofluorescence against endogenous β-Catenin was analysed via Operetta high-content microscope imaging. n = 3. DAPI served as nuclear marker. Identified known and new putative regulators of β-Catenin are highlighted. c Representative immunofluorescent images of endogenous β-Catenin (green) upon siRNA mediated knock-down of NTC (control), CTNNB1 and USP10, respectively. DAPI served as nuclear marker (blue). d Expression of CTNNB1 and USP10 in non-transformed (normal) and CRC (tumour) samples. Publicly available data from GEPIA. COAD (n = 275) and GTEx (n = 349) data were displayed as boxplots for USP10 and CTNNB1 expression. P-values were calculated using one-way ANOVA. Data was visualised using the online tool www.gepia.cancer-pku.cn. ***p < 0.001. e Correlation of gene expression between CTNNB1 and USP10 in human CRC. R: Spearman’s correlation coefficient. nT = 275, nN = 349. Data was visualised using the online tool www.gepia.cancer-pku.cn. f Publicly available patient survival data of CRC patients are stratified by relative expression of USP10. n = 206 (low) and n = 26 (high). Survival correlation analysis was performed using R2: Genomics Analysis and Visualization Platform, using the Tumour Colon - Smith dataset. g Representative images of immunohistochemistry (IHC) staining of a Tissue Micro Array (TMA) from CRC patients, comprising adjacent non-transformed tissue (adjacent nt) and CRC samples against β-Catenin and USP10. P-values were calculated using Mann–Whitney U test. *p < 0.05; **p < 0.005. h Immunoblotting of endogenous abundance of USP10, β-Catenin and MYC in non-transformed (WT) and patient matched CRC tumour samples (T) from two individual patients. β-Actin served as loading control. i Representative brightfield images of patient derived intestinal organoids, comprising either wild type (WT mucosa) or tumour derived organoids (CRC T5), respectively. Immunoblotting of endogenous abundance of USP10 and β-Catenin of patient organoids. β-Actin served as loading control. j Expression of USP10 in non-transformed (WT) and CRC patient derived organoids (tumour). Analysis was performed using R2: Genomics Analysis and Visualization Platform, using the Organoid - Clevers dataset. P-values were calculated using Mann–Whitney U test. ***p < 0.001.
Since USP10 was not implicated in intestinal homeostasis and β-Catenin signalling, next, we determined expression level of USP10 by interrogating publicly available patient data of colorectal cancer [25]. While USP10 was rarely mutated in CRC, it was predominantly upregulated, along with CTNNB1, when compared to adjacent wild-type tissue (Fig. 1d and Supplementary Fig. S1c), and USP10 and CTNNB1 demonstrated a significant degree of correlation of expression in CRC samples (Fig. 1e, nT = 275, nWT = 349, Spearman coefficient R = 0.79). Remarkably, elevated expression of USP10 in CRC is a strong indicator of overall poor patient survival in CRC (Fig. 1f), especially in the molecular subtypes CMS2-4 (Fig. S1e). Prompted by this observation, we next studied USP10 and β-Catenin levels by Immunohistochemistry (IHC) of tissue micro arrays (TMA) of CRC patients comprising non-transformed and tumour tissue. Not only was a difference in tissue architecture observed in CRC tissue samples, but USP10 and β-Catenin indeed showed a significant upregulation in CRC when compared to the adjacent tissue (Fig. 1g). Elevated protein abundance was furthermore analysed by using human samples from CRC resection surgeries, subjected to immunoblotting against endogenous USP10, β-Catenin and MYC. Non-transformed, adjacent tissue served as control. USP10, β-Catenin and the oncogene MYC were increased in tumour-samples compared to matched non-transformed tissue samples (Fig. 1h).
Publicly available data did highlight that expression of USP10 and CTNNB1 were elevated irrespective of CRC stage (Supplementary Fig. S1f). This was further validated using single cell sequencing data from two individual datasets, which demonstrated a tumour-specific increase in USP10 expression when compared to non-transformed, normal tissue (Supplementary Fig. S1g) as well in spatial transcriptomic data from a publicly available dataset (https://www.10xgenomics.com/datasets/visium-hd-cytassist-gene-expression-libraries-of-human-crc, Supplementary Fig. S1 extended). We tested this observation in human and murine intestinal wild type and tumour organoid models regarding the regulation of USP10 (Fig. 1i). Similar to the observation in patent-derived primary resected CRC tumours, the endogenous protein levels of USP10 was significantly increased in tumour derived organoids, compared to non-oncogenic (Fig. 1i). This was further supported by analysing publicly available expression data of patient derived non-oncogenic and CRC organoids [31] (Fig. 1j).
These data propose that USP10 is a novel regulator of β-Catenin stability, and a putative involvement of USP10 in WNT signalling, intestinal homeostasis and carcinogenesis.
Genetically engineered murine models of intestinal cancer demonstrate the upregulation of USP10 as an early event in CRC formationTo further investigate the expression of USP10 in the intestine, we examined weather USP10 is expressed in the intestinal stem cell niche and analysed unperturbed crypts (Fig. 2a, b and Supplementary Fig. S2a, b). Using fractionation of murine small intestine followed by immunoblotting we observed an enrichment of USP10 in crypts over villi (Fig. 2a). We confirmed that USP10 was abundant in intestinal crypts when compared to the villus (Fig. 2b and Supplementary Fig. S2a, b), and nuclear localised in intestinal stem cells using immunofluorescence (USP10+/β-Cateninnuclear; USP10+/Cd44high; USP10+/Lysozyme-; Fig. 2b and Supplementary Fig. S2b). This observation was further confirmed by analysing publicly available spatial transcriptomic data of murine intestine (https://www.10xgenomics.com/datasets/visium-hd-cytassist-gene-expression-libraries-of-mouse-intestine, Supplementary Fig. S2 extended).
Fig. 2: Genetically engineered murine models of intestinal cancer demonstrate the upregulation of USP10 as an early event in CRC formation.
a Schematic representation of murine small intestine and colon. Villi were scratched from the intestine and small intestinal and colonic crypts were isolated using EDTA. Isolated tissue from two individual mice was analysed for endogenous abundance of USP10, β-Catenin and Krt20. β-Actin served as loading control. (n = 2). b Representative immunofluorescent images of WT intestinal crypts of endogenous USP10 (green) and crypt cell specific markers. Upper panel: Lysozyme (magenta) marks Paneth cells. Lower panel: Cd44 (magenta) labels stem cells. DAPI served as nuclear marker (blue). White line indicates stretch of fluorescence quantification. Histogram of fluorescence over indicated length. c Schematic representation of acute in vivo CRC onset in wild type CD1 animals using colorectal instillation of lentivirus particles encoding sgRNA against murine Apc, targeting exon 10 (Apcex10), and constitutive expression of SpCas9. Viral backbone was pLenti-CRISPR-V2. pr.i. - pre infection. d Haematoxylin and eosin (H&E) staining of CRISPR mediated tumour onset in CD1 animals, 12 weeks post intracolonic instillation of virus. Insets highlight either non-transformed adjacent tissue or primary tumour upon Apc deletion. e Representative immunofluorescent images of mice shown in a and b of endogenous USP10 (green) and β-Catenin (red). DAPI served as nuclear marker (blue). Insets highlight either untransformed (1) or transformed (2) regions. Intensity of β-Catenin and USP10 staining was quantified using QuPath software. P-values were calculated using Mann–Whitney test. Individual cells/values are highlighted as dots. ***p < 0.001. f Insets from c. High magnification immunofluorescent images of intestines of CRISPR infected animals for USP10 (green) and β-Catenin (red). DAPI served as nuclear marker (blue). g Schematic representation of CRISPR-engineered murine CRC models using Adeno-associated viruses (AAV) to deliver sgRNA and HDR templates to either truncate endogenous APC within exon 9, Trp53 or point mutate endogenous Kras to KrasG12D. h Representative brightfield images of murine intestinal organoids, comprising either wild type (Cas9), after targeting and growth factor depleted selection upon CRISPR engineering of Apc exon 9 (Apcex9) and KRas to KRasG12D(AK), or upon co-deletion of Trp53 (APK), respectively. Immunoblotting of endogenous abundance of USP10 and β-Catenin in Cas9, AK and APK organoids. β-Actin served as loading control.
Intrigued by the expression of USP10 in unperturbed intestinal tissue, we tested whether the abundance of USP10 is enriched upon transformation of intestinal cells. First, we performed histopathologic analysis of individual tumours in animals, where carcinogenesis was induced either by acute CRISPR editing of Apc, causing a truncation at exon 10 (APCex10), or by loss of heterozygosity of Apc in a well-established mouse model of spontaneous intestinal cancer, Apcmin/+ (Fig. 2c–f, Supplementary Fig. S2c–f). Irrespective of genetic alteration of Apc, causal to carcinogenesis we observed that the protein level of USP10 was significantly upregulated in tumours within the GI tract, along with elevated protein levels of β-Catenin, when compared to non-transformed or non-oncogenic intestinal epithelium, respectively (Fig. 2e, f and Supplementary Fig. S2e, f).
Next, we wondered if discreet genetic alterations could be a contributor to USP10 upregulation. To address this question, we used murine wild type organoids (Cas9) and employed CRISPR gene editing to generate Apc:KrasG12D (AK) or Apc:Trp53:KrasG12D (APK) organoids (Fig. 2g, h and Supplementary Fig. S2g). Loss of Apc induced morphologic changes of wild type murine organoids and alleviated the requirement for WNT activating components in the growth medium. This is in accordance with previous reports, and likewise mutation of Kras to KrasG12D upregulated endogenous Erk1/2 signalling and alleviated the requirement for EGF supplementation (Supplementary Fig. S2g) [32]. In accordance the protein level of USP10 and β-Catenin was significantly enriched in transformed organoids when compared to parental control (Fig. 2h).
Thus, the upregulation of USP10 in colorectal cancer is an early event, caused by oncogenic transformation irrespective of genetic driver complexity, and coincides with elevated abundance of the WNT effector β-Catenin.
Truncation of APC allows for de novo protein-protein interaction between USP10 and β-Catenin in CRCWe hypothesized that β-Catenin directly interacts with USP10 in CRC and tested whether mutations within either β-Catenin or APC are a prerequisite to enable a protein-protein interaction. To this end, we co-immunoprecipitated endogenous USP10 and β-Catenin in the human CRC lines HCT116 (CTNNB1mutant/APCwildtype) and HT-29 (CTNNB1wildtype/APCtruncated) by either immunoprecipitating USP10 or β-Catenin first, followed by probing against the putative novel interaction partner (Fig. 3a, b). While USP10 and β-Catenin were singly immunoprecipitated in HCT116, no co-precipitation was observed. In contrast, USP10 co-immunoprecipitated with endogenous β-Catenin in HT-29 cells (Fig. 3a). This observation highlighted the possibility that the truncation status and length of APC is involved in the interaction between USP10 and β-Catenin. To examine this point, we used a panel of human CRC lines, comprising LS174T, DLD-1, SW480, SW620, Caco-2 and Colo320, which harbour varying truncation mutations within APC (Supplementary Fig. S3b, c). Endogenous co-immunoprecipitation of USP10 and β-Catenin only occurred in Colo320, confirming that a proximal truncation in APC is a prerequisite, as cell lines carrying distal deletions, such as DLD-1, Caco-2 and SW480/SW620, failed to co-immunoprecipitate USP10 with β-Catenin (Supplementary Fig. S3b, c). The truncation status within APC could have potential therapeutic implications, since analysing patient survival data and stratification of CRC patients regarding APC status (truncation within first 1000 amino acids or after, or carrying point mutations), indicated a trend towards shorter survival for short APC variant carriers (Supplementary Fig. S3a).
Fig. 3: Truncation of APC allows for de novo protein-protein interaction between USP10 and β-Catenin in CRC.
a Representative input and endogenous co-immunoprecipitation of USP10 and β-Catenin in human CRC cell lines either wild type for APC, HCT116APCwt, or truncated HT-29APCmut. IgG served as antibody specificity control. β -Actin served as loading control. Input represents 3% of total loading. n = 3. b Schematic representation of truncating mutations reported in the APC gene in the CRC cell lines HCT116 and HT-29. Dark blue box = 15 AAR domains, green small boxes = 20 AAR domains, large green boxes = SAMP domains. 15- and 20-AAR = β-Catenin amino acid repeats; SAMP = Axin binding sites. Images adapted from the publicly available database www.uniprot.org. c Schematic model of acute truncation of APC at amino acid 867 in HCT116 via CRISPR gene editing. d Bargraph of proximity ligation assay (PLA) between USP10 and β-Catenin in either APC wild type (APCwt) or APC867 truncated (APCmut) HCT116. Data analysed from more than 750 cells over two independent experiments per condition. P-values were calculated using Mann–Whitney U test. Representative immunofluorescent images of endogenous USP10 (green), β-Catenin (red) and the corresponding PLA (mustard) in either APCwt of APCmut HCT116. DAPI served as nuclear marker. n = 2. e Schematic overview of the in vitro binding assay in µSPOT format. Intrinsically disordered regions of USP7, USP10 and USP36, respectively, were determined by <50 pLDDT score in their individual AlphaFold2 structural prediction and represented as 15 mer peptides overlapping 12 or 11 amino acids. For binding assays, µSPOT slides bearing the peptide library were incubated with recombinantly expressed and purified β-Catenin. β-Catenin binding to the on-chip peptides was detected by immunostaining with a chemiluminescent readout. f Overview of binding intensities of recombinant β-Catenin towards discreet unstructured regions of USP7, USP10 and USP36. Gaps indicate the presence of structured domains within the DUBs. Colour code indicates binding intensity. Peptides with the globally highest binding intensity (N-terminal region of USP10 residues 7–21) are underlined in green and represented as a bar graph in panel g). Mean of n = 3. g Identified amino acid sequence within the N-terminal unstructured part of USP10 binding to recombinant β-Catenin. Bar graph shows binding intensity (abs. = absolute intensity). Mean of n = 3 with corresponding standard deviation. h Full positional scan of the most prominent β-Catenin binding hotspot USP107-21 identified in the overlapping scan (panel g). Each residue of the peptide sequence was systematically varied to every other proteogenic amino acid and their β-Catenin binding intensities are shown relative to the wildtype sequence. Note that amino acid variations for certain positions result in drastic reductions in binding intensity compared to the wildtype sequence, thus suggesting direct interactions of the respective sidechains with β-Catenin. Mean of n = 3.
Furthermore, based on the genetic alterations reported for HT-29 and Colo320, we concluded that the 15- and 20 β-Catenin binding amino acid Armadillo repeats (AAR) domains within APC are required to directly compete for binding of USP10 to β-Catenin. Hence, we postulate that the putative de novo interaction between USP10 and β-Catenin requires the loss of the AAR domains. Proximity ligation assays (PLA) between USP10 and β-Catenin in either control or CRISPR mediated APC-truncated HCT116 cells confirmed that APC competed with USP10 for binding to β-Catenin (Fig. 3c, d).
To further interrogate the interaction and map the USP10 binding site required for β-Catenin interaction, we conducted a µSPOT protein binding assay [33]. Here, the intrinsically disordered regions (IDR) of USP10, along with the IDR sequences of a known β-Catenin binder, USP7 [13], or USP36 (an additional DUB comprised of large unstructured regions), were displayed as overlapping peptide libraries and probed with recombinant β-Catenin (Fig. 3e). We identified USP10 residues 7QYIFGDFSPDEFNQF21 (Fig. 3f–h) to mediate direct and robust binding to β-Catenin. Intriguingly, by assessing the binding affinity of β-Catenin towards its known interactors Axin1, APC or TCF4 we found that the presence of the USP10 peptide did interfere with binding of β-Catenin to AXIN1 and APC, pointing towards a high affinity of β-Catenin towards USP10 and TCF4 (Supplementary Fig. S3d, e). Co-immunoprecipitation of endogenous β-Catenin in HT-29 transiently transduced with either control, wild type USP10 or USP10 depleted for amino acids 7–21 (USP10Δ7-21) revealed that the point mutant had a reduced interaction potential with endogenous β-Catenin (Supplementary Fig. S3f). This was further studied using AlphaFold2 MultimerV1.0 (AF2M) [34], with the complete sequences of USP10 and β-Catenin as input, predicts the same residues within USP10 to engage with β-Catenin, as identified by µSPOT protein binding assay (Supplementary Fig. S3g, h). Remarkably, this binding site is overlapping with APC and AXIN1 binding to β-Catenin (Supplementary Fig. S3f, g).
Taken together, we discovered a direct USP10-β-Catenin interaction as well as that both USP10 with APC compete for the same β-Catenin binding site. Thus, lending a molecular explanation for the observed indirect β-Catenin stabilizing effect of APC truncations.
Acute deletion of USP10 in intestinal stem cells of D. melanogaster rescues hyperplasia and lethality of the ApcQ8/Q8 modelAs USP10 and the entire Wnt pathway is highly conserved between species we used D. melanogaster to investigate its involvement in intestinal homeostasis and hyperproliferation upon loss of function mutations within APC, [35] (Similarity in aa: 379/821 - (46%), Identity in aa: 254/821 - (30%); Gaps: in aa:185/821 - (22%) https://www.flyrnai.org/cgi-bin/DRSC_prot_align.pl?geneid1 = 38103&geneid2 = 9100; Fig. 4 and Supplementary Fig. S4). Firstly, we assessed the impact of shRNA-mediated elimination of dUSP10 on intestinal progenitor homeostasis. Intestinal progenitor cells were marked by GFP expression, driven under the control of the escargot regulatory region (esg::GAL > GFP), and immunofluorescence against armadillo, the fly ortholog to β-Catenin, that is expressed in intestinal stem cells (ISCs, Supplementary Fig. S4a). Expression of shRNA against USP10 resulted in a marked reduction of ISCs when compared to a LacZ control shRNA (Supplementary Fig. S4a, b).
Fig. 4: Acute deletion of USP10 in intestinal stem cells of D.melanogaster rescues hyperplasia and lethality of the ApcQ8/Q8 mutant flies.
a Representative immunofluorescence of fly midguts. ApcQ8/+ heterozygotes are highly similar to wildtype midguts (not shown). Midguts of homozygous ApcQ8 mutants exhibit hyperproliferation of ISC (positive for the intestinal stem cell marker Delta (red)). Elimination of USP10 using USP10 inverted repeats (UAS-IR) suppresses the progenitor hyperproliferation phenotype observed in midguts of homozygous ApcQ8 mutants. b Quantification of total stem cell abundance in all three conditions. Significance as compared to “esg > +; ApcQ8” was calculated using one-way ANOVA. **p < 0.005; ***p < 0.001. c qRT-PCR analysis of the expression of USP10, armadillo and escargot in midguts isolated from either ApcQ8 or ApcQ8USP10KD flies. mRNA was normalised to Actb. Error bars represent standard deviation of 3 biological replicates. d Kaplan–Meier plot of adult survival of the indicated genotypes. ApcQ8n = 24, ApcQ8:esg-USP10in = 17.
Moreover, we tested for a genetic interaction between APC truncation and USP10 in a tumour-like setting using the ApcQ8 hyperplasia model [36]. The allele ApcQ8 harbours a premature stop codon leading to a significant truncation of Apc and loss of the β-catenin binding sites. Immunofluorescent analysis of D. melanogaster midguts revealed that heterozygous loss of Apc had a minor effect on overall tissue homeostasis highly similar to wildtype midguts. In contrast, midguts derived from adult animals carrying a homozygous LOF truncating mutation within Apc (ApcQ8/Q8) presented an entirely disorganised intestine (Fig. 4a). This midgut was robustly populated by escargot-positive progenitors, many of them expressing the stem cell marker and Notch ligand Delta. (Fig. 4a, b). Transcriptional analysis of midguts isolated from ApcQ8/Q8 revealed a significant increase in dUSP10 mRNA levels (Supplementary Fig. S4e). This is in alignment with the identified expression pattern observed in human and murine CRC and stresses a high degree of mechanistic similarities between the chosen model organisms. Knockdown of dUSP10, however, suppressed the stem cell and progenitor expansion observed in homozygous ApcQ8, and animals presented a midgut resembling a normal appearance (Fig. 4a, b). Analysis of isolated midguts from either ApcQ8/+, ApcQ8/Q8or ApcQ8/Q8 flies expressing an shRNA against dUSP10 (USP10i;ApcQ8/Q8) in intestinal stem cells indicated a significant reduction in overall USP10 transcript abundance, along with reduced expression of the stem cell marker escargot (Fig. 4c and Supplementary Fig. S4e).
Lastly, we investigated the impact of dUSP10 deletion on overall survival in the background of Apc-truncation driven hyperplasia model. While the survival of heterozygous ApcQ8/wt flies was similar to wildtype files, homozygous ApcQ8/Q8 mutation were characterized with a temperature-sensitive lethality (Fig. 4d and Supplementary Fig. S4f). Strikingly, expression of an shRNA against dUSP10 in intestinal progenitors (USP10i;ApcQ8/Q8) restored longevity, likely by negating the adverse effects on overall tissue homeostasis and growth imprinted by ApcQ8/Q8 (Fig. 4d).
These data demonstrate an epistatic genetic linkage between USP10 and truncated APC that is required for ectopic stem cell proliferation.
USP10, via controlling β-Catenin protein stability, regulates WNT signalling and stemness signature genesTo further elucidate the function of USP10 in CRC in the context of APC truncation, we deleted endogenous USP10 by co-targeting of exon 2 and 10 in HT-29 and HCT116, respectively (Fig. 5a and Supplementary Fig. S5a, b). Depletion of USP10 in HT-29 resulted in a marked reduction of β-Catenin, along with reduction in the CRC protein marker and WNT target gene LGR5 (Fig. 5a, b). Loss of USP10 enhanced overall ubiquitylation of β-Catenin (Fig. 5c) and accelerated protein turnover in HT-29 cells (Fig. 5d and quantified in 5e). Depletion of USP10 in HCT116 a cell line that harbour non-truncated Apc, however, had no effect on overall β-Catenin abundance nor ubiquitylation (Supplementary Fig. S5b–d), confirming the dependency of the USP10-β-Catenin interaction on APC-truncation. Interestingly, while cells deleted for USP10 by CRISPR mediated targeting did show reduced abundance and increased ubiquitylation of β-Catenin, longitudinal propagation of HT-29ΔUSP10 was not possible. Targeted cells within a heterogeneous cell pool were rapidly outcompeted by wildtype cells (Supplementary Fig. S5e). This is in line with previous reports of cell lethality upon USP10 loss [37]. To by-pass this long-term lethality, we used an inducible knock down system, comprising two independent shRNA against USP10, to acutely deplete the DUB in HT-29 (Supplementary Fig. S5f). USP10 depleted HT-29 showed a significantly reduced proliferation, when compared to control vector transduced cells (Supplementary Fig. S5g).
Fig. 5: USP10 regulates WNT signalling and stemness signature genes via controlling β-Catenin protein stability.
a Immunoblot against endogenous USP10, β-Catenin and LGR5 in APC mutant HT-29 cells upon CRISPR mediated depletion of USP10. Two different cell pools (USP10−1 and USP10−2) along with non-targeting control (ctrl) cells are shown. β-Actin served as loading control. n = 3. b Quantitative RT-PCR of USP10, CTNNB1 and LGR5 expression of HT-29 USP10 CRISPR pool (USP10−2) compared to control (ctrl) cells. Error bars represent standard deviation of n = 3 independent experiments. Significance was calculated using Student’s t test. **p < 0.005; ***p < 0.001. n.s. non-significant. c Tandem Ubiquitin Binding Entity (TUBE) assay of endogenous poly-ubiquitylated proteins, followed by immunoblotting against endogenous β-Catenin in HT-29 USP10 CRISPR cells (USP10−2). Immunoblot against endogenous USP10 is shown. β-Actin served as loading control. n = 2. d Cycloheximide (CHX) chase assay (100 μg/ml) of control (shNTC) or shUSP10-2 expressing HT-29 cells for indicated time points. Representative immunoblot analysis of USP10 and β-Catenin. β-Actin served as loading control. n = 3. e Quantification of relative protein abundance of β-Catenin, normalised to β-Actin, as shown in d. Significance was calculated using Student’s t test. n = 3 *p < 0.05; ***p < 0.001. f Representative immunoblot against endogenous USP10 and β-Catenin in APC mutant HT-29 cells upon DOX-inducible overexpression of GFP control (GFP), catalytical active GFP-USP10 (GFP USP10WT) and a catalytical inactive mutant of USP10 (GFP USP10CA). β-Actin served as loading control. (n = 3). g Quantitative RT-PCR of USP10, CTNNB1 and KRT20 expression of HT-29 cells overexpressing exogenous USP10. Error bars represent standard deviation of n = 3 independent experiments. Significance was calculated using Student’s t test. **p < 0.005; ***p < 0.001. n.s. non-significant. h Growth-curve of GFP USP10WT and GFP USP10CA overexpressing HT-29 cells compared to GFP control cells. Error bars represent standard deviation of n = 3 independent experiments. Significance was calculated using one-way ANOVA. ***p < 0.001. n.s. non-significant. i Representative immunofluorescence images of conditional USP10WT and USP10CA overexpression and GFP control in HT-29 cells. J Quantification of i. Mean intensity over well was measured and normalised to GFP control. Error bars represent standard deviation of n = 3. Significance was calculated using unpaired t-test. *p < 0.05; ***p < 0.001; n.s. non-significant.
A stem cell niche-specific contribution of USP10 was further supported by analysing the whole proteome of HT-29 cells treated with either non-targeting (ctrl) or USP10 siRNA for 24 h (Supplementary Fig. S5h, i). Among the downregulated proteins were proteins associated with the stem cell niche, including TCF4 (TCF7L2), TNFRSF21, NOTCH2, LGR4, CD44, along with reduced protein level of the proto-oncogene MYC, a direct target of WNT signalling (Supplementary Fig. S5h).
To investigate the extent of regulation of the WNT effector β-Catenin by USP10, and using a gain-of-function approach, we conditionally overexpressed either wild type (USP10WT) or a catalytic inactive variant of USP10 (USP10CA) in the CRC line HT-29. Conditional increase in USP10 led to an increase in β-Catenin abundance on protein as well as mRNA level (Fig. 5f, g). The catalytic activity of USP10 is required to facilitate these effects on β-Catenin, as USP10CA failed to stabilise β-Catenin (Fig. 5f, g). Expression of USP10 significantly enhanced overall proliferation of HT-29 cells, when compared to vector or catalytic inactive mutant control cells (Fig. 5h). Given that β-Catenin directly controls intestinal homeostasis and stem- and cancer cell maintenance, next, we tested if USP10 affects the expression of essential stem- and CRC pathways. Immunofluorescence imaging of HT-29 expressing either USP10WT or USP10CA demonstrated that proteins associated with the CSC stem niche, such as β-Catenin, OLFM4, LGR5, ASCL2 or CD44 were significantly upregulated in a USP10WT dependent fashion (Fig. 5i, j).
These observations establish that USP10 regulates the ubiquitylation and abundance of β-Catenin in an APC truncation dependent manner, promoting the expression of WNT pathway and (cancer) stem cell signatures and CRC growth.
USP10 is required to maintain CRC cell identity, stemness and tumour growthTo investigate the clinical relevance and dependency of human CRC tumours towards USP10 in a patient-relevant setting, we used patient-derived organoids (Fig. 6a). The patient organoid line P6T carries mutations comparable to HT-29; a truncating mutation resulting in a short APC variant (R876*) and a longer variant (P1420fs), making it a suitable candidate to test USP10 dependency. 3 weeks post infection and selection with either a non-targeting control shRNA (shNTC) or an shRNA targeting USP10, patient-derived organoids were analysed (Fig. 6b–h). Loss of USP10 significantly reduced overall organoid numbers and size (Fig. 6c, d). Transcriptomic analysis of P6TshNTC and P6TshUSP10 organoids revealed that USP10 is involved in the regulation of WNT signalling, differentiation and stem cell maintenance (Fig. 6f, g). Stem cell-related genes, such as LGR5, LEF1, AXIN2 or LRIG1 were reduced upon loss of USP10, while the expression of differentiation associated genes, such as MUC2 or KRT20, were enriched (Fig. 6f). Furthermore, loss of USP10 led to enriched gene sets associated with stress signalling, such as unfolded protein response and reactive oxygen species signalling in P6T tumour organoids (Fig. 6h). These observations are in line with the results obtained from HT-29 cells and clearly demonstrate that USP10 is involved in the maintenance/propagation of a pro-tumorigenic signature, supporting stem cell-like features of cells expressing high levels of USP10 that is required for the tumorigenic state.
Fig. 6: USP10 is required to maintain CRC cell identity, stemness and tumour growth.
a Schematic overview of workflow for isolation, characterisation and silencing of USP10 in patient derived CRC organoid P6T (Oncode Organoid bank). b Representative brightfield images of stable transformed human P6T organoids infected with either shRNA against USP10 or with a non-targeting control. n = 10 field of view. Highlighted are individual and intact organoids. c Quantification of relative organoid number (per field of view) one week post infection with either a control (shNTC) or shUSP10. Statistical analysis was performed using unpaired t test. p < 0.0001. Images were quantified using QuPath (version0.4.2) and ImageJ (FIJI). Boxplots were generated using Graphpad Prism8. In box plots, the centre line reflects the median and the upper and lower box limits indicate the first and third quartiles. Whiskers extend 1.5× the IQR. P-values were calculated using Mann–Whitney U test. ***p < 0.0001. d Quantification of relative organoid size (per field of view) one week post infection with either a control (shNTC) or shUSP10. Statistical analysis was performed using unpaired t test. p < 0.0001. Images were quantified using QuPath (version0.4.2) and ImageJ (FIJI). Violinplots were generated using Graphpad Prism8. P-values were calculated using Mann–Whitney U test. ***p < 0.0001. e Volcano-plot of differential expressed genes upon knock-down of USP10 in human P6T organoids, relative to expression in shNTC infected control organoids. Significantly regulated genes are highlighted in red, respectively. USP10 is highlighted. n = 3. f Heatmaps showing expression of genes linked to WNT signalling, differentiation and NOTUM signalling in either shNTC or shUSP10 P6T organoids. n = 3. g, h GSEA analysis of P6T organoids expressing an shRNA sequence targeting USP10 or non-targeting control (shNTC). Changes in gene expression were analysed and enrichment plots for gene sets mapping to WNT signalling, EMT, UPR and ROS are shown. i Representative brightfield images of stable transformed murine Aex9PKG12D organoids (APK9). Two different shRNAs against USP10 and shNTC expressing organoids were generated. j Representative immunoblot of USP10 and β-Catenin protein upon shRNA mediated knock-down of endogenous USP10. β-Actin served as loading control. Quantification was calculated from n = 3. k Gene set enrichment analysis of MsigDB gene sets, deregulated in shUSP10-1 compared to shNTC APK9 organoids. l Gene set enrichment analysis of intestinal specific
留言 (0)