
記住我

The protocol for this study is reported based on the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 Checklist [33]: defining standard protocol items for clinical trials (Additional file 1). The study has been approved by the Ethics Committee of Heping Hospital Affiliated to Changzhi Medical College (approval number 2023 No.029) and has been registered in the Chinese Clinical Trial Registry (ChiCTR) (registration number ChiCTR2300078275). This study is still ongoing.
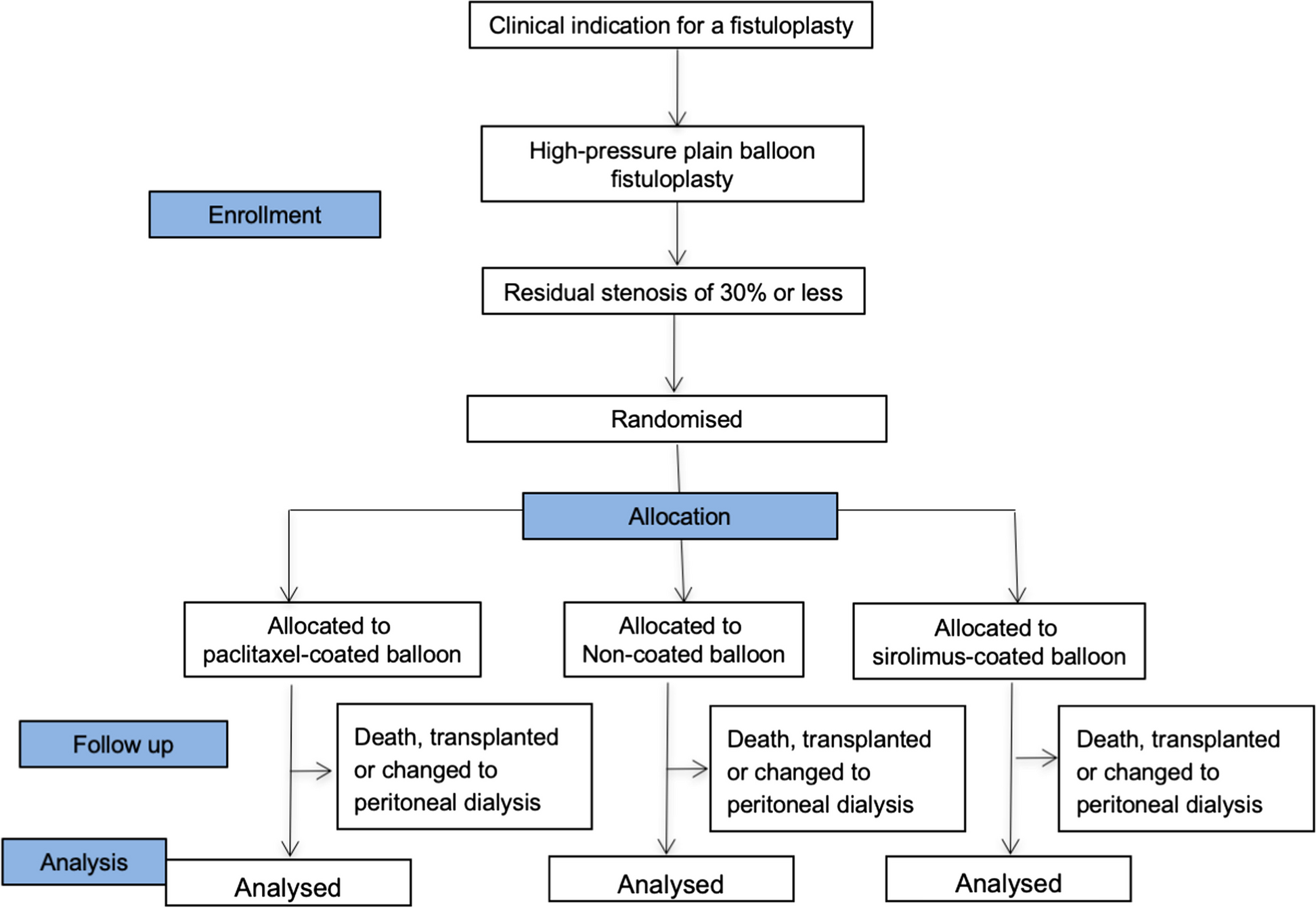
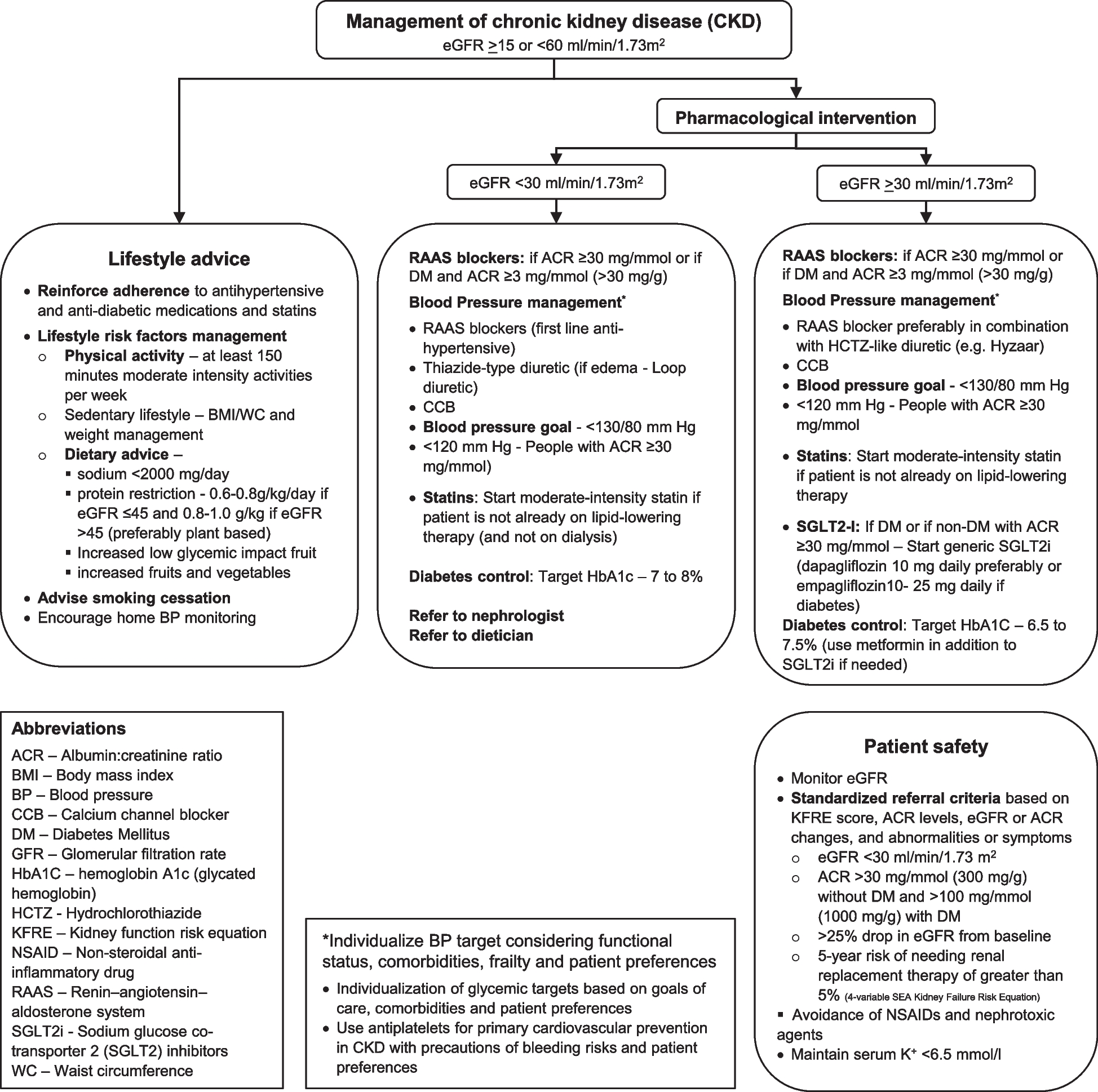
Trial designThis study is a prospective dose-finding clinical trial using up-and-down method with biased-coin design (BCD) and determines the EC90 of remifentanil inhibiting cardiovascular responses to tracheal intubation during Ai-guided ciprofol anesthesia (Figs. 1 and 2). Based on previous study [21], a total of 60 patients scheduled to undergo elective surgery will be recruited in this study. Data analysis will be performed according to the superiority principle. The study will be carried out at Heping Hospital Affiliated to Changzhi Medical College and will be conducted according to the principles of the Helsinki Declaration (2000 edition, Edinburgh). Written informed consent will be obtained from each patient or, if the patient cannot provide informed consent, from the surrogate of the patient.
Fig. 1
Flow chart of the study design
Fig. 2
Standard Protocol Items: Recommendations for Interventional Trials (adapted from SPIRIT figure). MOAA/S, modified observer’s assessment of alertness/sedation score; Ai, depth of anesthesia index
The study will continue for 12 months, and all the selected individuals will be from hospitalized elective surgery patients with tracheal intubation during general anesthesia. The researchers will conduct screening in accordance with the established criteria. Data collection will start from the collection of basic data collection and continue until the end of follow-up (Fig. 2).
Randomization and implementationThis study is an adaptive dose-finding trial with biased-coin up-and-down sequential allocation design. In adaptive clinical trials with BC-UDM, randomization is often not used in the traditional sense. Instead of randomly assigning participants to different treatment groups, this design adapts the treatment allocation based on the outcomes observed during the trial. The next patient’s treatment assignment may depend on the response of the previous patient, creating a sequence of adaptive treatment allocations. While this design is not based on randomization in the conventional sense, it still aims to minimize bias and ensure the validity of the study results by adjusting the treatment allocation based on observed responses.
The random walk rules for the sequential allocation of dose levels to patients in this trial were conducted using the BCD-UDM [21, 33,34,35,36]. Patients are sequentially assigned the next higher, same, or next lower dose level according to the probability distribution of BCD-UDM, which is determined by ethical considerations as well as the patient’s binary endpoints [21].
Jianing Guo generated the random allocation sequence, Luoyun Li and Zeru Zhang enrolled participants and assigned participants to interventions, and analysis was done by Fangsheng Xu who was blinded to the interventions.
Study participants and recruitmentWe will recruit 60 patients scheduled to undergo elective surgery. These patients will be recruited from Heping Hospital Affiliated to Changzhi Medical College after they meet the eligibility criteria and sign their informed consent. We plan to enroll the first patient on July 1, 2024 and to end on December 31, 2024. All participants will sign the informed consent form for participating in the clinical trials.
On the day before surgery (or Friday for patients undergoing surgery next Monday), researchers authorized by the chief investigator will examine the list of patients scheduled for surgery and their medical records to determine potential participants based on our inclusion and exclusion criteria. Then, they will visit these patients and formally invite them to participate. For patients who meet the inclusion/exclusion criteria and receive written informed consent, baseline data will be collected, including demographic data, preoperative diagnosis, medical history, medication history, and surgical history, as well as the main results of physical examinations and laboratory and instrument examinations.
Inclusion criteriaInclusion criteria of participants in this trial is based on a previous study [22,23,24,25,26]. Inclusion criteria are as follows: (1) patients of general anesthesia for elective surgery; (2) age 18 to 64 years old; (3) ASA is graded I–II; and (4) body mass index (BMI) 18–28 kg/m2.
Exclusion criteriaExclusion criteria are as follows: (1) allergies or contraindications to opioids, ciprofol, propofol, and their components; (2) use of other sedatives such as ciprofol or propofol or midazolam within 24 h before surgery; (3) patients with severe central nervous system, respiratory or circulatory system diseases; respiratory diseases, difficult airway, liver dysfunction, renal dysfunction, mental disorders, long-term use of psychotropic drugs, and cognitive dysfunction, long-term use of psychotropic or sedative-hypnotic drugs, drug abuse and drinking; (4) patients with Allen test positive, hypertension, hemodynamic instability (systolic blood pressure [SBP] < 90 mmHg or > 180 mmHg, diastolic blood pressure [DBP] > 110 mmHg, peripheral blood oxygen saturation [SpO2] < 90%); (5) participation in other clinical studies within recent 1 month; and (6) patients with a history of difficult endotracheal intubation or suspected difficult endotracheal intubation, defined as a Mallampati class IV airway; retrognathia; restricted neck movements; or more than two criteria among the following: Mallampati class III airway, mouth opening less than 35 mm, or thyromental distance less than 65 mm. All of these parameters were estimated by an experienced anesthesiologist.
Discharge criteriaThe discharge criteria are as follows: (1) individuals are required to withdraw during the trial period; (2) violation of trial procedures; and (3) the occurrence of serious adverse events (AEs).
InterventionIn this study, we will investigate the EC90 of remifentanil blunting cardiovascular responses to tracheal intubation during Ai-guided ciprofol anesthesia using BC-UDM.
Definition of binary endpointThe remifentanil during anesthesia induction with ciprofol inhibits cardiovascular responses to tracheal intubation and is simplified to a binary endpoint (i.e., positive/negative). Patients’ systolic blood pressure (SBP), diastolic blood pressure (DBP), mean arterial pressure (MAP), heart rate (HR), and Ai values were recorded before induction, at baseline (defined as the average of 3 and 1 min measured values before tracheal intubation), and 1 and 3 min after tracheal intubation. The increase in the MAP or HR was the difference between the average of the 1 and 3 min measured values after tracheal intubation and its baseline value. Based on previous study [22,23,24,25,26], if mean MAP or HR was elevated by 15% of the value compared with the baseline values before intubation, a positive response was defined, and a negative response was defined as unaltered or elevated mean MAP or HR by < 15%.
The probability of negative response must maintain the same direction of change (increasing or decreasing) with increasing dose. We assume that it is increasing and denote the relationship between dose and negative-response probabilities by the function F(x), where x is the dose magnitude variable (Fig. 3).
Fig. 3
Diagram of dose-response curve. EC90, 90% effective concentration; EC50, median effective concentration; 90% CI, 90% confidence intervals
Dose allocation and dose spacingRemifentanil during anesthesia induction with ciprofol inhibits cardiovascular responses to tracheal intubation and must be ordered as a discrete set of increasing doses of the same treatment drug. Preferably, the allowed doses are uniformly spaced in an algebraic sequence.
In previous literature, there were differences in initial dose and dose spacing [22,23,24,25,26]. Based on previous study [22,23,24,25,26], the initial dose and dose spacing of remifentanil in this study were 3.5 ng/ml and 0.5 ng/ml, respectively. Remifentanil was administered at an effect-site concentration of 3.5 ng/ml to the first patient. The target effect-site concentration of remifentanil employs a dose range based on known clinical effectiveness and is split into 8 to 12 dose levels [21]. Dose range is 3.0 to 8.0 ng/ml, including 11 levels: 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, and 8.0 ng/ml.
Dose-transition and titration rulesThe dose-transition rules are based on the doses and responses of the last patient or several patients rather than on all patient data going back to the beginning of the experiment. When EC90 was to be determined (τ = 0.9), the following formula was used: probability (B) = (1 − τ)/τ = (1 − 0.9)/0.9 = 0.1/0.9 ≈ 0.11, where B is the target percentage [21, 37, 38].
Dose titration can be weighted on the probability of positive or negative response. If the cardiovascular response to tracheal intubation was a positive response, the effect-site concentration (Ce) of remifentanil would increase by 0.5 ng/ml. Conversely, if a patient was negative response based on cardiovascular responses to tracheal intubation, the next patients either received the same Ce (probability 1–11% = 89%) or a lower 0.5 ng/ml Ce (probability 11%), which was randomly decided using a computer-generated random list prepared by a statistician who was not involved in any other part of the study.
According to BC-UDM rules [21, 37, 38], doses are allocated to patients sequentially and only allow for increasing the dose by one level, decreasing by one level, or repeating the same dose. The dose-transition rules are based on the doses and responses of the last patient or several patients rather than on all patient data going back to the beginning of the trials. Furthermore, the rules do not use any estimated quantity that changes during the study.
Stopping rulesIt is suggested in previous simulation study that including at least 50–60 patients will provide stable estimates of the target dose for EC90 [20, 21]. This trial will be recruiting 60 cases for evaluating EC90 of remifentanil blunting cardiovascular responses to tracheal intubation during Ai-guided ciprofol anesthesia using BC-UDM.
Anesthesia implementation and managementPreparation before general anesthesiaPrior to the surgery, all patients were routinely fasted of food and water without any premedication. A standard monitoring and anesthetic technique was applied to all patients in the operating room. After entering the operating room, one of the arms was inserted with a venous catheter of 20- or 22-gauge by a nurse, and a total of 10 ml/kg Ringer’s solution was administered before anesthesia induction for fluid expansion. Patients with a negative Allen test were subjected to ultrasound-guided invasive arterial puncture and catheterization under local anesthesia by an anesthesiologist, along with real-time monitoring of arterial blood pressure.
During the perioperative period, electrocardiograms (ECG), pulse oximetry (SpO2), heart rate (HR), invasive blood pressure (IBP), and end-tidal carbon dioxide (PetCO2) were monitored (BeneVision M15 Monitor, Mindray, China), and Ai is also continuously monitored using a monitor for the depth of anesthesia (ConView YY-106; Pearlcare Medical Technology Company Limited, Zhejiang, China).
In order to prevent intraoperative awareness, the modified observational alertness/sedation assessment (MOAA/S) score (qualitative evaluation) and Ai (quantitative evaluation) were used for sedation evaluation in this study. MOAA/S score is divided into 0–5 levels; each level score represents the different clinical levels of sedation [39] (Table 1). Ai is a better parameter to estimate alterations in consciousness. As a new monitoring index of anesthesia depth, Ai is based on the sample entropy of the electroencephalogram and then obtains a dimensionless value of 0–99 through certain calculation methods [31, 32]. As the optimal anesthesia depth in clinical practice, the Ai value is controlled between 40 and 60, and the MOAA/S is less than or equal to 1 (Table 1).
Table 1 Assessment of sedation levelAnesthesia inductionThe order of anesthesia induction is as follows: remifentanil-ciprofol-rocuronium bromide (Fig. 2). Before induction of intravenous anesthesia, preoxygenation of patients with 100% oxygen via facial masks for 3 min is applied. Anesthesia is performed using TCI for administering remifentanil, which starts at a predetermined target effect-site concentration and is administered through an infusion pump (Fresenius Kabi, France) according to the Minto model [40]. After reaching equilibrium between remifentanil plasma and effect-site concentrations, an intravenous injection of ciprofol (0.4 mg/kg) [30] is administered within 30 s. When the patients is unconsciousness (MOAA/S scores ≤ 1 and Ai ≤ 60), rocuronium (0.6 mg/kg) was given within 30 s, and artificial ventilation was initiated.
A laryngoscopy and endotracheal intubation is performed 2 min after rocuronium injections, using a unified visual laryngoscope (TD-C-IV-3, Zhejiang Youyi Medical Equipment Co. Ltd, China) and an ordinary endotracheal tube (CGPO, TUORen, Henan Province, China); the diameter of the tube was individualized by the patient’s height and gender.
All endotracheal intubations were performed by one experienced attending anesthesiologist; those patients whose endotracheal intubation was not successful at one time or whose intubation time was longer than 1 min were excluded from the study. General anesthesia was maintained using 0.8 MAC sevoflurane with oxygen (1 L/min), and end-tidal carbon dioxide concentrations were maintained at 35–45 mmHg using mechanical ventilation. Three minutes after endotracheal intubation, remifentanil’s effect-site concentration remained unchanged.
During anesthesia induction, Ai and MOAA/S scores were also continuously monitored and recorded. MOAA/S score is assessed by the anesthesiologists every 10 s after administration of 0.4 mg/kg ciprofol until three consecutive MOAA/S scores ≤ 1 and Ai ≤ 60. Ai is quite the same as BIS does.
During data collection, excessive hemodynamic changes include systolic blood pressure < 80 or > 180 mmHg and HR < 50 bpm or > 120 bpm. Hypoxia is defined as SpO2 ≤ 92% for more than 10 s. If the patient experiences excessive hemodynamic changes, hypoxemia, severe muscle tremors, or persistent chest wall stiffness, we will handle it according to the emergency disposal plan, and the patient will withdraw from this study. The following cases will be treated with the same concentration of remifentanil.
Outcomes assessmentPrimary outcomeThe primary outcomes were Ce of remifentanil inhibiting cardiovascular responses to tracheal intubation during Ai-guided ciprofol anesthesia using BCD-UDM in 90% of the study population.
Secondary outcomesFollowing the EC90 calculation, the data were further analyzed for secondary outcomes to compare those who were positive/negative response for tracheal intubation. Secondary outcomes of this study will include the following: (1) the changes of the hemodynamic indices (SBP or HR) and indices derived from Ai during tracheal intubation; (2) AEs related to remifentanil combined with ciprofol anesthesia, include great hemodynamic change, hypoxemia, muscle tremor, symptoms of chest wall rigidity, choking cough, and postoperative nausea and vomiting; and (3) the changes of MOAA/S score and Ai values during anesthesia induction.
Statistical analysisSample size estimationA sample size of at least n = 50 to 60 for the EC90 was determined according to a statistical reference and previous recommendation [21, 37] and will provide stable estimates of the target dose for the most realistic scenarios. Therefore, 60 patients were enrolled in this study.
Data analysesStatistical analysis was performed using SPSS version 25.0 software (IBM, Armonk, NY, USA). Normally distributed continuous variables are described as mean ± standard deviation (SD), while nonnormally distributed continuous variables are described as the median and interquartile range (IQR). Categorical variables are described as numbers (percentages).
EC90 was calculated using centered isotonic regression with a bias-corrected Morris 90% confidence interval (CI) derived by bootstrapping using “dosefind” and “quickinverse” commands in the Centered Isotonic Regression R package (R’s “cir” package authored and maintained by Dr. Oron) [21, 37]. Pooled-adjacent-violators algorithm (PAVA)-adjusted response rates were estimated using the weighted isotonic regression method [21].
Biased-coin up-and-down trials generate binary (positive/negative) response data. The proportion of negative responses at each dose is calculated and plotted by a dose-response plot. Targeting the EC90, from the dose-response observation pairs, isotonic regression methods are used to estimate the dose-response curve [21].
Data collection and monitoringThe statistical professionals are responsible for formulating the statistical analysis plan through consultation with the main researchers, establishing the database, and using the SPSS statistical analysis system for statistics. A comprehensive efficacy analysis was conducted in accordance with the program set, and the whole analysis set, demographic and other baseline characteristics, and other efficacy indicators were analyzed in accordance with the program set.
The Data Monitoring Committee (DMC) consists of a doctor in charge of data collection and sorting, a scientific research manager, and a statistician. The DMC will meet three times a year throughout the entire research process. The DMC is responsible for safeguarding the interests of trial participants, evaluating the safety of intervention measures, and supervising the overall progress of the trial. Any deviation from the protocol will be recorded in the report. All major plan modifications will be communicated to relevant parties and updated in the trial register.
The project team designed and prepared the experiment and will announce the results. The group will hold monthly meetings to discuss the progress of the research. The doctor from the DMC will record the actual number of individuals enrolled, the cases of exclusion, demographic and other baseline characteristics, compliance analysis, safety analysis, incidence of complications and combined treatment, and comprehensive efficacy evaluation. The demographic characteristics, medical history, and treatment history of the patients will be described. The scientific research management committee will have access to the final trial dataset. At the end of the study, the original data and results will be submitted to the scientific research management committee; they will be disclosed to the public after the results are published.
Safety evaluationAE refers to the appearance or progression of any discomfort, syndrome, or disease symptoms that occur during clinical trials, which can affect the patient’s health. Any abnormalities in clinical trials indicating the presence of disease and/or organ toxicity, as well as severe abnormalities that require active treatment (such as discontinuation of medication, increased follow-up, and diagnostic studies), are considered AEs.
During clinical research, researchers should fill in the AE record form truthfully and in detail, recording the clinical manifestations, occurrence time, severity, duration, measures taken, and outcomes of AEs.
When an AE occurs, the observing doctor can decide whether to suspend observation based on the situation. All AEs should be tracked and recorded in detail until the patient’s situation is properly resolved or the patient is in a stable state. If laboratory testing is abnormal and clinically significant, it should be traced back to the pre-treatment levels.
Patient and public involvementPatients with scheduled for elective surgery were involved during our previous pilot trial and reviewed project for the present study. At the protocol design stage, we gained opinions from participating medical center on the content of ethics and safety evaluation.
Dissemination planThe results of this study will be presented at anesthesia conferences (local and international). The main investigation results will be reported at the trial registration office. The complete research report will be submitted for publication in anesthesia journals, preferably open-access journals.
留言 (0)